博文
HiCPro分析流程详解
|||
本篇接着上一篇《HiCPro的安装与使用》,详细讲解Hi-C数据比对软件HiCPro的分析流程。HiCPro的安装与使用,请查看:
http://blog.sciencenet.cn/blog-2970729-1182259.html
3. HiCPro分析流程
HiCPro处理各步骤流程如下,总体来说可以分为两大部分,对应HiCPro的两个Steps:step1比对,step2 Hi-C fragment相关分析
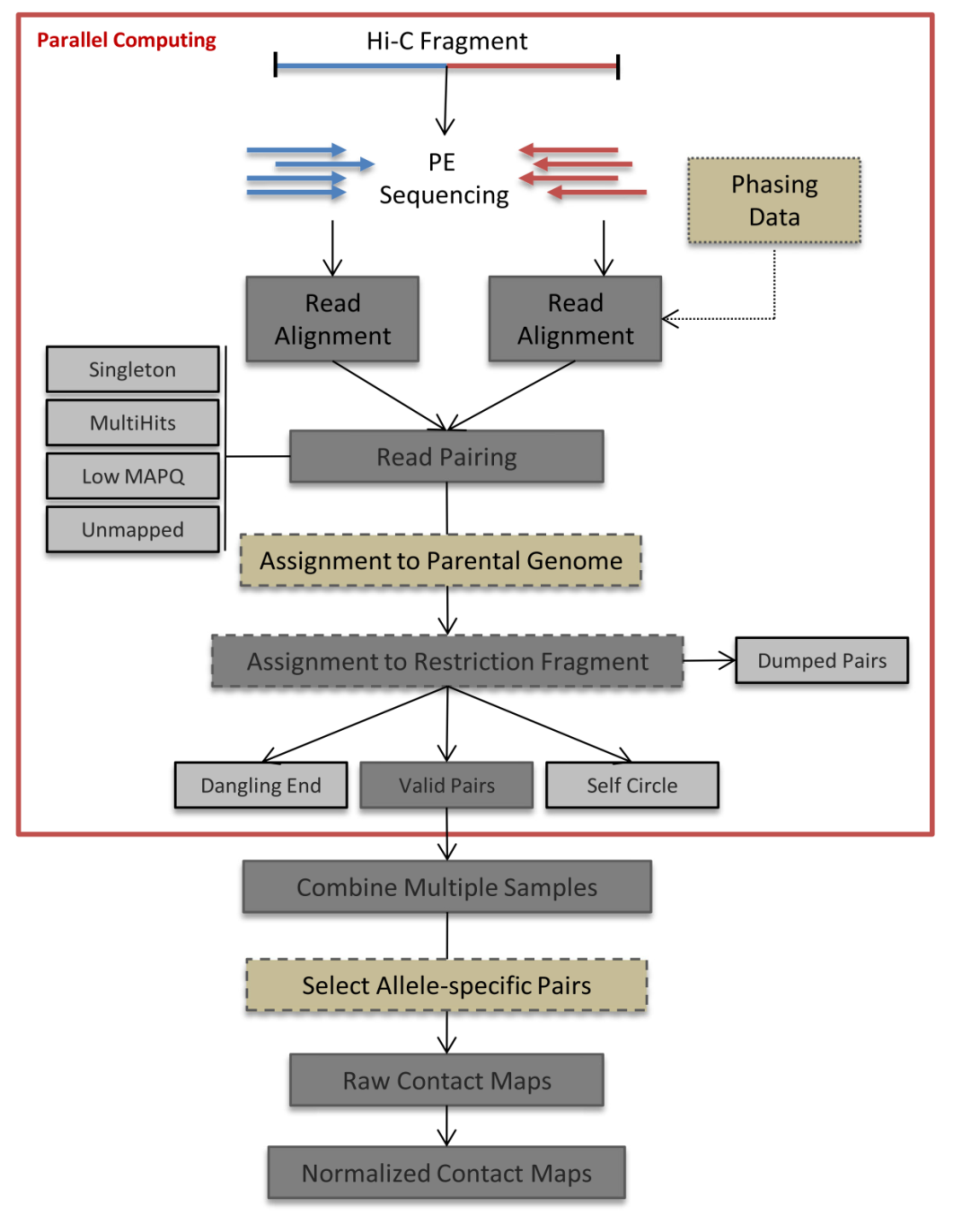
3.1 HiCPro先采用bowtie分别对PE reads进行比对
R1 reads比对:
/path/to/bowtie2 --very-sensitive -L 30 --score-min L,-0.6,-0.2 --end-to-end --reorder --un bowtie_results/bwt2_global/sample/sample_R1_sample_genome_ref.bwt2glob.unmap.fastq --rg-id BMG --rg SM:sample_R1 -p 24 -x /path/to/ref/index -U rawdata/sample/sample_R1.fastq.gz 2>> logs/sample/sample_R1_bowtie2.log| /path/to/samtools view -F 4 -bS - > bowtie_results/bwt2_global/sample/sample_R2_sample_genome_ref.bwt2glob.bam
## bowtie2相关参数如下
# 注意:这里使用--very-sensitive。采用bowtiew的Hi-C 比对软件通常会用最严格比对
# bowtie2 -L选项是seed substrings的长度,3-32之间
# bowtie2 --end-to-end是entire read must align; no clipping
# bowtie2 --reorder是force SAM output order to match order of input reads
# bowtie2 --un选项表示输出未比对上的fastq序列到该文件中
# bowtie2 --rg-id & --rg指定group id相关信息
# bowtie2 -p指定线程数
# bowtie2 -x指定参考基因组索引
# bowtie2 -U指定输入的单端reads文件
## samtools view相关参数
-F 4 清理unmapped read。因为未比对上的reads已经记录在unmap.fastq文件中。输出的bam文件中不会再保留未比对上的alignment信息,这样可以减少后续读取文件的速度。
R2 reads比对参数与R1一致
/path/to/bowtie2 --very-sensitive -L 30 --score-min L,-0.6,-0.2 --end-to-end --reorder --un bowtie_results/bwt2_global/sample/sample_R2_sample_genome_ref.bwt2glob.unmap.fastq --rg-id BMG --rg SM:sample_R2 -p 24 -x /path/to/ref/index -U rawdata/sample/sample_R2.fastq.gz 2>> logs/sample/sample_R2_bowtie2.log| /path/to/samtools view -F 4 -bS - > bowtie_results/bwt2_global/sample/sample_R2_sample_genome_ref.bwt2glob.bam
完成后会在bowtie_results目录下生成一个bwt2_global的文件夹,该文件夹下还有一个以样本名命名的子目录,其中包括R1比对bam文件,unmapped fastq文件,R2的比对bam文件,unmapped fastq文件
3.2 对未比对上的reads进行trim和再比对
# 对R1未比对上的reads进行trim
/path/to/HiC-Pro_2.11.1/scripts/cutsite_trimming --fastq bowtie_results/bwt2_global/sample/sample_R1_sample_genome_ref.bwt2glob.unmap.fastq --cutsite GATCGATC --out bowtie_results/bwt2_local/sample/sample_R1_sample_genome_ref.bwt2glob.unmap_trimmed.fastq > logs/sample/sample_R1_sample_genome_ref.bwt2glob.unmap_readsTrimming.log 2>&1
# --fastq指定输入的未比对上的reads
# --cutsite指定酶切位点序列
# --out输出trim之后的reads
# 对R2未比对上的reads进行trim。参数与R1一致
/path/to/HiC-Pro_2.11.1/scripts/cutsite_trimming --fastq bowtie_results/bwt2_global/sample/sample_R2_sample_genome_ref.bwt2glob.unmap.fastq --cutsite GATCGATC --out bowtie_results/bwt2_local/sample/sample_R2_sample_genome_ref.bwt2glob.unmap_trimmed.fastq > logs/sample/sample_R2_sample_genome_ref.bwt2glob.unmap_readsTrimming.log 2>&1
# 对R1 trimmed reads进行再比对
注意:这一步与第一次比对(即3.1)节中的比对过程差异是:
samtools view 没有用-F 4,即将未比对上的reads也记录在了bam文件中
这是因为后期将会统计比对率,需考虑到unmapped reads
/path/to/bowtie2 --very-sensitive -L 20 --score-min L,-0.6,-0.2 --end-to-end --reorder --rg-id BML --rg SM:sample_R1_sample_genome_ref.bwt2glob.unmap -p 24 -x /path/to/ref/index -U bowtie_results/bwt2_local/sample/sample_R1_sample_genome_ref.bwt2glob.unmap_trimmed.fastq 2>> logs/sample/sample_R1_sample_genome_ref.bwt2glob.unmap_bowtie2.log | /path/to/samtools view -bS - > bowtie_results/bwt2_local/sample/sample_R1_sample_genome_ref.bwt2glob.unmap_bwt2loc.bam
# 对R2 trimmed reads进行再比对
/path/to/bowtie2 --very-sensitive -L 20 --score-min L,-0.6,-0.2 --end-to-end --reorder --rg-id BML --rg SM:sample_R2_sample_genome_ref.bwt2glob.unmap -p 24 -x /path/to/ref/index -U bowtie_results/bwt2_local/sample/sample_R2_sample_genome_ref.bwt2glob.unmap_trimmed.fastq 2>> logs/sample/sample_R2_sample_genome_ref.bwt2glob.unmap_bowtie2.log | /path/to/samtools view -bS - > bowtie_results/bwt2_local/sample/sample_R2_sample_genome_ref.bwt2glob.unmap_bwt2loc.bam
运行完成后会在bowtie_results目录下生成一个bwt2_local的文件夹,该文件夹下还有一个以样本名命名的子目录,其中会生成R1 trimmed reads比对的bam结果,以及trim后还是无法比对上的reads序列文件。R2的也有类似的两个文件。
3.3 分别对R1 & R2 reads两次比对的结果合并
# 对R1两次比对的结果合并
/path/to/samtools merge -@ 24 -n -f bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.bam bowtie_results/bwt2_global/sample/sample_R1_sample_genome_ref.0.bwt2glob.bam bowtie_results/bwt2_local/sample/sample_R1_sample_genome_ref.bwt2glob.unmap_bwt2loc.bam
# -f选项,表示如果存在输出文件则overwrite
# -n表示Input files are sorted by read name
# 对R2两次比对的结果合并
/path/to/samtools merge -@ 24 -n -f bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.bam bowtie_results/bwt2_global/sample/sample_R2_sample_genome_ref.bwt2glob.bam bowtie_results/bwt2_local/sample/sample_R2_sample_genome_ref.bwt2glob.unmap_bwt2loc.bam
# 对R1合并后的结果排序
/path/to/samtools sort -@ 24 -n -T tmp/sample_R1_sample_genome_ref -o bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.sorted.bam bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.bam
# 对R2合并后的结果排序
/path/to/samtools sort -@ 24 -n -T tmp/sample_R2_sample_genome_ref -o bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.sorted.bam bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.bam
# 将R1输出的.sort.bam文件改名为.bam文件,这一步估计是程序写完后发现有个bug,为了不改动后面的程序,加的一步
mv bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.sorted.bam bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.bam
# 将R2输出的.sort.bam文件改名为.bam文件
mv bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.sorted.bam bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.bam
运行完成后,会在bowtie_results目录下生成一个bwt2文件夹,和以样本名为名字的子文件夹。在其中生成两个以bwt2merged.bam结尾的文件,分别代表R1 & R2的结果
3.4 统计比对率
采用的方法是samtools view -c,具体如下:
# 分别统计R1 & R2最终结果中总的reads和成对的reads
/path/to/samtools view -c bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.bam
/path/to/samtools view -c bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.bam
/path/to/samtools view -c -F 4 bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.bam
/path/to/samtools view -c -F 4 bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.bam
# 分别统计R1 & R2两次比对结果中,比对上的reads数
/path/to/samtools view -c -F 4 bowtie_results/bwt2_global/sample/sample_R1_sample_genome_ref.bwt2glob.bam
/path/to/samtools view -c -F 4 bowtie_results/bwt2_global/sample/sample_R2_sample_genome_ref.bwt2glob.bam
/path/to/samtools view -c -F 4 bowtie_results/bwt2_local/sample/sample_R1_sample_genome_ref.bwt2glob.unmap_bwt2loc.bam
/path/to/samtools view -c -F 4 bowtie_results/bwt2_local/sample/sample_R2_sample_genome_ref.bwt2glob.unmap_bwt2loc.bam
3.5 采用HiCPro的mergeSAM.py程序合并PE reads
/path/to/python /path/to/HiC-Pro_2.11.1/scripts/mergeSAM.py -q 10 -t -v -f bowtie_results/bwt2/sample/sample_R1_sample_genome_ref.bwt2merged.bam -r bowtie_results/bwt2/sample/sample_R2_sample_genome_ref.bwt2merged.bam -o bowtie_results/bwt2/sample/sample_sample_genome_ref.bwt2pairs.bam
# -q指定最小的mapping quality
# -t表示需生成比对统计结果
# -v用于debug
# -f/--forward & -r/--reverse分别输入上一步生成的PE reads分开比对最终的bam文件(即3.3中结果)
# -o输出处理后的结果
此外,HiCPro的mergeSAM.py程序还有以下几个参数:
[-s/--single] <report singleton> 给出单端比对的结果
[-m/--multi] <report multiple hits> 给出多处比对的结果
运行完成后,会在bwt2的子目录中生成后缀为bwt2pairs.bam的过滤结果文件。以及其统计文件(后缀为.bwt2pairs.pairstat的文件),其中会统计完整的比对信息。包括Total_pairs_processed, Unmapped_pairs, Low_qual_pairs, Unique_paired_alignments, Multiple_pairs_alignments, Pairs_with_singleton, Low_qual_singleton, Unique_singleton_alignments, Multiple_singleton_alignments, Reported_pairs
至此,比对的部分全部完成,所有的结果都在bowtie_results目录下
=====================================================
再进行Hi-C fragment相关分析,所有的结果都在hic_results目录下
3.6 利用HiCPro的mapped_2hic_fragments.py程序将比对结果转化为Hi-C片段信息
/path/to/python /path/to/HiC-Pro_2.11.1/scripts/mapped_2hic_fragments.py -v -a -f /path/to/restriction_enzyme_cutting_site.MboI.txt -r bowtie_results/bwt2/sample/sample_sample_genome_ref.bwt2pairs.bam -o hic_results/data/sample
# -v用于debug
# -a记录所有的信息,包括self-circle, dangling end等
# -f指定最开始时,检测出的基因组序列上的酶切位点信息文件,即HiC-Pro_2.11.1/bin/utils/digest_genome.py生成的结果
# -r指定bowtie2比对的最终结果,即3.5节中的结果
# -o指定输出目录
此处,还可以指定insert size的阈值等,具体可参见--help
再对输出的valid pairs文件进行排序:
LANG=en; sort -T tmp -k2,2V -k3,3n -k5,5V -k6,6n -o hic_results/data/sample/sample_sample_genome_ref.bwt2pairs.validPairs hic_results/data/sample/sample_sample_genome_ref.bwt2pairs.validPairs
# 这里是直接将valid paris原路径排序
3.7 对所有的valid pairs进行合并,并且去掉PCR duplication
LANG=en; sort -T tmp -S 50% -k2,2V -k3,3n -k5,5V -k6,6n -m hic_results/data//sample/sample_sample_genome_ref.bwt2pairs.validPairs | awk -F"\t" 'BEGIN{c1=0;c2=0;s1=0;s2=0}(c1!=$2 || c2!=$5 || s1!=$3 || s2!=$6){print;c1=$2;c2=$5;s1=$3;s2=$6}' > hic_results/data/sample/sample.allValidPairs
这一步是先将多个valid pairs文件进行合并,例如加测了几次,如果之前没有合并,这里可以合到一起。然后再确定当前行和上一行是否相同,如果相同则为PCR duplication,需去除
3.6和3.7节的结果都生成在hic_results/data目录中
3.8 采用HiCPro的merge_statfiles.py程序对bowtie2比对的多个统计结果合并
/path/to/python /path/to/HiC-Pro_2.11.1/scripts/merge_statfiles.py -d bowtie_results/bwt2/sample/ -p *_R1*.mapstat -v > hic_results/stats/sample/sample_R1.mmapstat
/path/to/python /path/to/HiC-Pro_2.11.1/scripts/merge_statfiles.py -d bowtie_results/bwt2/sample/ -p *_R2*.mapstat -v > hic_results/stats/sample/sample_R2.mmapstat
/path/to/python /path/to/HiC-Pro_2.11.1/scripts/merge_statfiles.py -d bowtie_results/bwt2/sample/ -p "*.pairstat" -v> hic_results/stats/sample/sample.mpairstat
/path/to/python /path/to/HiC-Pro_2.11.1/scripts/merge_statfiles.py -d hic_results/data//sample/ -p "*.RSstat" -v> hic_results/stats/sample/sample.mRSstat
# 这一步统计的结果都在hic_results/stats目录下
3.9 跟据BIN_SIZE来构建matrix
这一步会按照HiCPro config-hicpro.txt文件中指定的BIN_SIZE,对valid pairs进行分配,构建matrix
cat hic_results/data//sample/sample.allValidPairs | /path/to/HiC-Pro_2.11.1/scripts/build_matrix --matrix-format upper --binsize 20000 --chrsizes /path/to/ref/reference.size --ifile /dev/stdin --oprefix hic_results/matrix/sample/raw/20000/sample_${bsize}
# --chrsizes是最初统计出的基因组中每条序列的长度
运行完,各分辨率的matrix输出到hic_results/matrix/sample/raw目录下
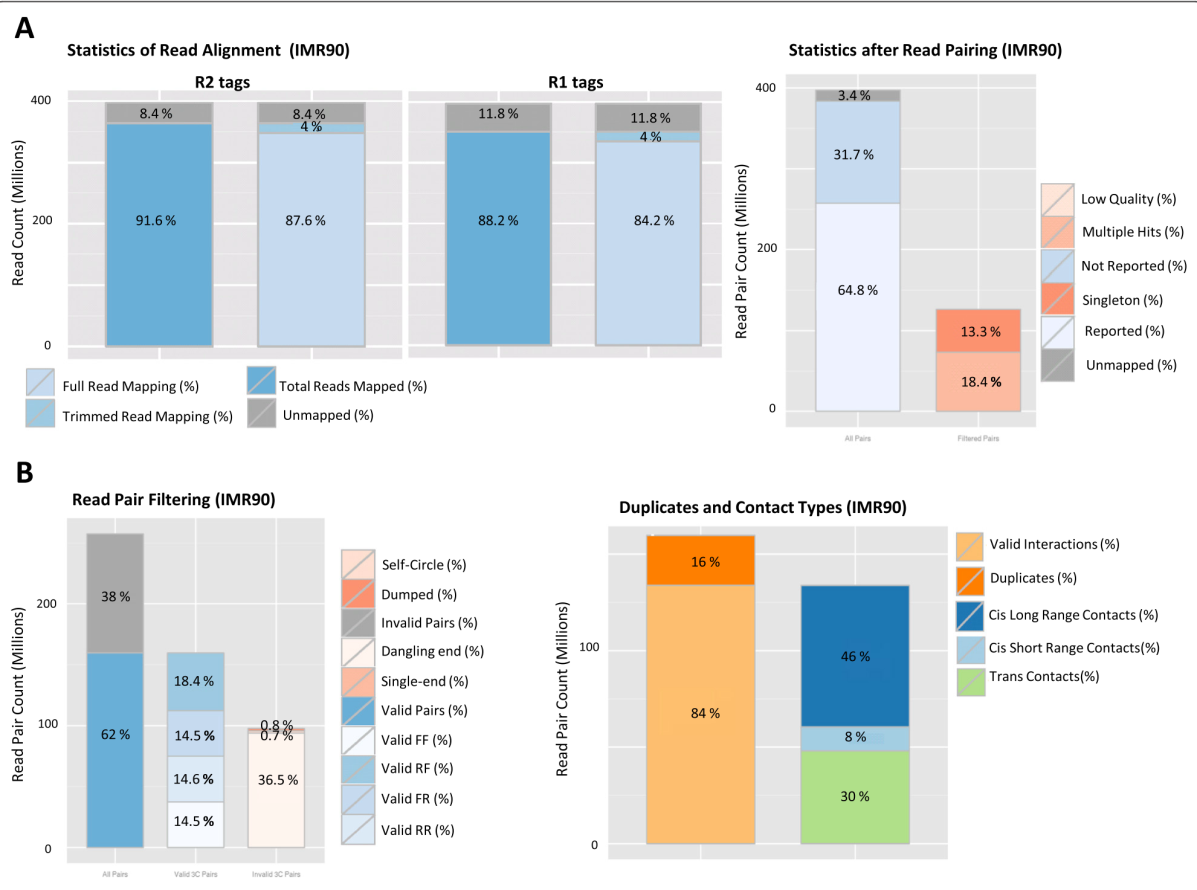
3.10 对统计结果画图
会生成5个图,分别是plotHiCContactRanges_sample.pdf, plotHiCFragmentSize_sample.pdf, plotMappingPairing_sample.pdfplot,HiCFragment_sample.pdf, plotMapping_chrysanthemum.pdf, 如下图:
3.11 采用ice对raw matrix做normalization
/path/to/python /path/to/HiC-Pro_2.11.1/scripts/ice --results_filename hic_results/matrix/sample/iced/20000/sample_20000_iced.matrix --filter_low_counts_perc 0.02 --filter_high_counts_perc 0 --max_iter 100--eps 0.1 --remove-all-zeros-loci --output-bias 1 --verbose 1 hic_results/matrix//sample/raw/20000/sample_20000.matrix
# 对各分辨率生成的raw matrix做normalization,结果都输出在hic_results/matrix/sample/ice目录下
https://blog.sciencenet.cn/blog-2970729-1185463.html
上一篇:Hi-C数据比对软件HiCPro的安装与使用
下一篇:HiCPro安装中的iced问题