І©ОД
GWAS·ЦОцЦРSNPҪвКН°Щ·ЦұИPVE | өЪЛДЖӘЈ¬MLMДЈРНЦРИзәОКЦ¶ҜјЖЛгPVEЈҝ
||
ЎёПөБРІҝ·ЦЈәЎ№
GWAS·ЦОцЦРSNPҪвКН°Щ·ЦұИPVE | өЪТ»ЖӘЈ¬SNPҪвКН°Щ·ЦұИЦ®әНОӘәОҙуУЪ1Јҝ
GWAS·ЦОцЦРSNPҪвКН°Щ·ЦұИPVE | өЪ¶юЖӘЈ¬GLMДЈРНЦРИзәОјЖЛгPVEЈҝ
GWAS·ЦОцЦРSNPҪвКН°Щ·ЦұИPVE | өЪИэЖӘЈ¬MLMДЈРНЦРИзәОјЖЛгPVEЈҝ
ҪсМмҪйЙЬТ»ПВИзәОКЦ¶ҜјЖЛгMLMДЈРНGWASөДPVEҪб№ыЎЈТтОӘGAPITЦРөДMLMДЈРНУЦPVEҪб№ыЈ¬ө«КЗіЈУГөДGEMMAЎўGCTAөДGWASҪб№ыІўГ»УРPVEЈ¬ұҫЖӘҪйЙЬТ»ПВИзәОёщҫЭGWASҪб№ыКЦ¶ҜјЖЛгЈ¬УГRУпСФҪшРРСЭКҫЎЈ
1. ІОҝјОДПЧ
КЧПИКЗХвёцВЫМіөДДЪИЭЈәHow to determine the percent phenotypic variation explained (PVE) by a selected SNP?
❝Feofanova, Elena. (2020). Re: How to determine the percent phenotypic variation explained (PVE) by a selected SNP?. Retrieved from: https://www.researchgate.net/post/How_to_determine_the_percent_phenotypic_variation_explained_PVE_by_a_selected_SNP/5e1600dd661123743209bc17/citation/download.
❞
АпГжҪйЙЬБЛјЖЛг·Ҫ·ЁЈә
 ЖдЦРІОҝјөДОДПЧКЗЈә
ЖдЦРІОҝјөДОДПЧКЗЈә
❝Shim, H., Chasman, D.I., Smith, J.D., Mora, S., Ridker, P.M., Nickerson, D.A., Krauss, R.M., and Stephens, M. (2015). A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One 10, e0120758.
❞
АпГжөДёҪјю1ЈәSupplementary Information: S1: Computing proportion of variance in phenotype explained by a given SNP (PVE).
Хыёц№«КҪИзПВЈә
 ЧоәуЈ¬НкХыөД№«КҪИзПВЈә
ЧоәуЈ¬НкХыөД№«КҪИзПВЈә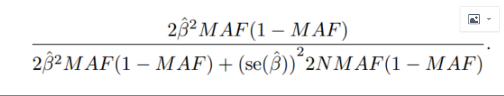 ЖдЦРЈә
ЖдЦРЈә
ОӘGWASЦРөДeffectЦө MAF ОӘSNPөДMAFҙОөИО»»щТтЖөВК ОӘGWASЦРeffectЦөөДұкЧјОуЈЁseЈ© N ОӘGWASЦРёГSNPІОУл·ЦОцөДёцМеКэ
2. GAPITЦРMLMДЈРН·ЦОцPVEЦө
gaipitЦРөДMLMДЈРНҙъВлИзПВЈә
# GWAS ·ЦОц
library(data.table)
source("http://zzlab.net/GAPIT/GAPIT.library.R")
source("http://zzlab.net/GAPIT/gapit_functions.txt")
myGd = fread("mdp_numeric.txt",header=T,data.table = F)
myGM = fread("mdp_SNP_information.txt",header = T,data.table=F)
myY = fread("dat_plink.txt",data.table = F)
head(myY)
covar = fread("cov_plink.txt",data.table = F)[,-1]
names(covar)[1] = "Taxa"
head(covar)
myGAPIT = GAPIT(Y = myY[,c(1,3)],GD = myGd, GM = myGM,
# PCA.total=3,
CV = covar,Random.model=TRUE,
model="MLM")¶ФҪб№ыҪшРРҙҰАнЈ¬јЖЛгPVEЦөЈ¬Ҫб№ыИзПВЈә
a2 = fread("05_lmm_gapit/GAPIT.MLM.V3.GWAS.Results.csv") %>% arrange(P.value) # GAPIT MLM
a2$PVE = a2$Rsquare.of.Model.with.SNP - a2$Rsquare.of.Model.without.SNP
head(a2)
a2 = a2 %>% select(1,lmm_effect = effect,maf = maf,lmm_p.value = P.value,
lmm_FDR_p = `FDR_Adjusted_P-values`,
lmm_Rsquare_with_snp = Rsquare.of.Model.with.SNP,
lmm_Rsquare_without_snp = Rsquare.of.Model.without.SNP,
lmm_Rs = Rs)
head(a2)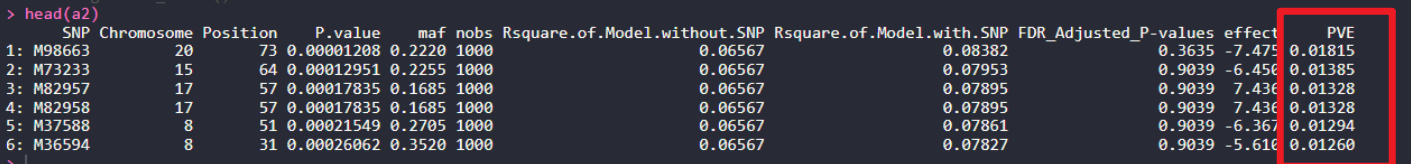 ХвАпЈ¬GWASҪб№ыЦРЈ¬ТтОӘГ»УРeffectөДseөДЦөЈ¬ЛщТФОЮ·ЁКЦ¶ҜФЛЛгЈ¬ПВГжОТГЗҝҙТ»ПВGEMMAәНGCTAөДfast-GWAЈ¬УГН¬СщөДКэҫЭЈ¬ҪшРРGWAS·ЦОцЈ¬ІўКЦ¶ҜјЖЛгPVEЦөЈ¬әНGAPITЦРөДMLMДЈРНөДPVEЦөҪшРР¶ФұИЎЈ
ХвАпЈ¬GWASҪб№ыЦРЈ¬ТтОӘГ»УРeffectөДseөДЦөЈ¬ЛщТФОЮ·ЁКЦ¶ҜФЛЛгЈ¬ПВГжОТГЗҝҙТ»ПВGEMMAәНGCTAөДfast-GWAЈ¬УГН¬СщөДКэҫЭЈ¬ҪшРРGWAS·ЦОцЈ¬ІўКЦ¶ҜјЖЛгPVEЦөЈ¬әНGAPITЦРөДMLMДЈРНөДPVEЦөҪшРР¶ФұИЎЈ
3. GEMMAҪшРРMLMДЈРНөДGWAS·ЦОц
GEMMAҪшРРGWAS·ЦОцЈ¬·ЦОӘБҪІҪЈә
өЪТ»ІҪЈә№№ҪЁGҫШХу өЪ¶юІҝЈәҪшРРMLMөДGWAS·ЦОц
# №№ҪЁGҫШХу gemma-0.98.1-linux-static -bfile ../geno/b -gk 2 -p p1.txt # ҪшРРLMM·ЦОц gemma-0.98.1-linux-static -bfile ../geno/b -k output/result.sXX.txt -lmm 1 -p p1.txt -c cov.txt
Ҫб№ыИзПВЈә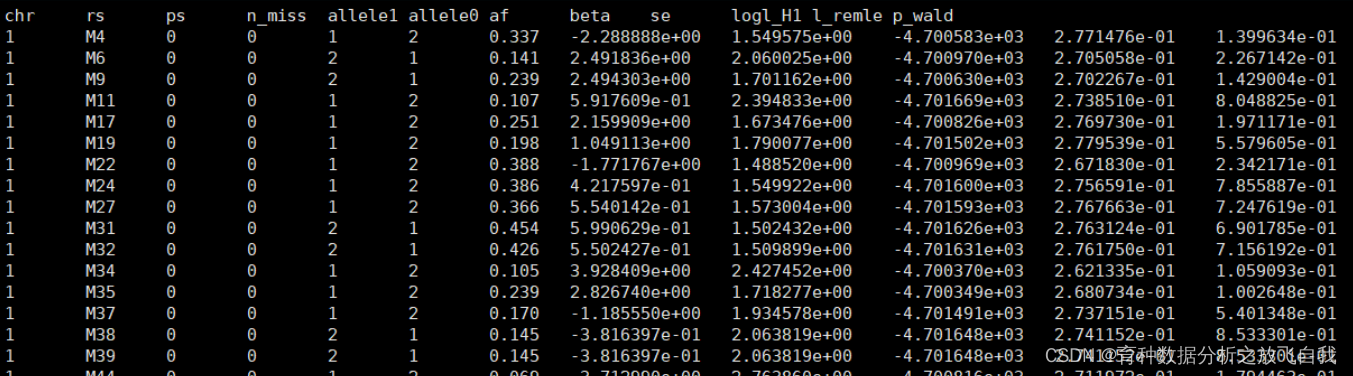 Ҫб№ыЦРЈә
Ҫб№ыЦРЈә
betaОӘeffect seОӘse p_wald ОӘPЦө n_miss ОӘЧЬёцМеКэөДИұК§Ј¬nОӘЧЬёцМеКэјхИҘИұК§ afОӘmafҙОөИО»»щТтЖөВК
ЛщТФЙПГжҪб№ыЈ¬¶БөҪRУпСФЦРЈ¬УГПВГ湫КҪҪшРРјЖЛгPVEЈә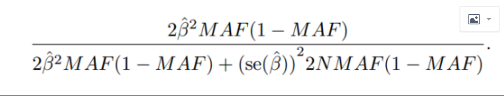 ХвАпөДNОӘ1000Ј¬јЖЛгҪб№ыИзПВЈә
ХвАпөДNОӘ1000Ј¬јЖЛгҪб№ыИзПВЈә
a4$pve = (2*(a4$beta^2*a4$af*(1-a4$af)))/(2*a4$beta*a4$af*(1-a4$af) + a4$se^2*2*1000*a4$af*(1-a4$af)) head(a4)
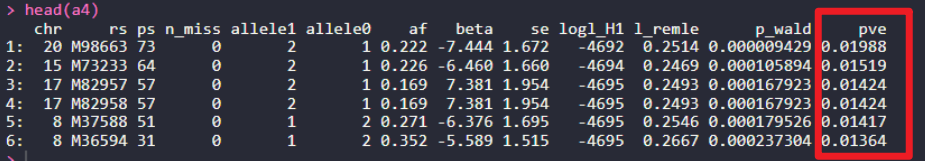
ұИҪПGAPITөДMLMДЈРНөДPVEәНКЦ¶ҜёщҫЭGEMMAөДMLMјЖЛгөДPVEҪб№ыЈ¬ҝЙТФҝҙөҪSNPM98663Ј¬ЛьөДPVEФЪGAPITЦРКЗ0.01815Ј¬ФЪGEMMAЦРКЗ0.01988Ј¬Ҫб№ыУРР©ІоТмЈ¬ПВГжОТГЗҝҙТ»ПВПа№ШПөКэЎЈ
ЎёұИҪППа№ШПөКэЈәЎ№
re = merge(a2,a4,by.x="SNP",by.y = "rs") head(re) re %>% select(P.value,p_wald) %>% cor re %>% select(effect,beta) %>% cor re %>% select(PVE,pve) %>% cor re %>% select(PVE,pve) %>% plot
GAPITәНGEMMAөДPЦөұИҪПҪб№ыЈә0.9986Ј¬»щұҫТ»ЦВЎЈ
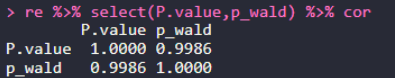
GAPITәНGEMMAөДeffectЦөұИҪПҪб№ыЈә0.9996Ј¬»щұҫТ»ЦВЎЈ
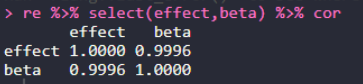 GAPITәНGEMMAөДPVEЦөұИҪПҪб№ыЈә0.9991Ј¬»щұҫТ»ЦВЎЈ
GAPITәНGEMMAөДPVEЦөұИҪПҪб№ыЈә0.9991Ј¬»щұҫТ»ЦВЎЈ
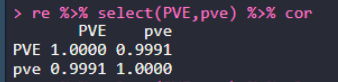 БҪҝоИнјюөДPVEөДЙўөгНјЈә
БҪҝоИнјюөДPVEөДЙўөгНјЈә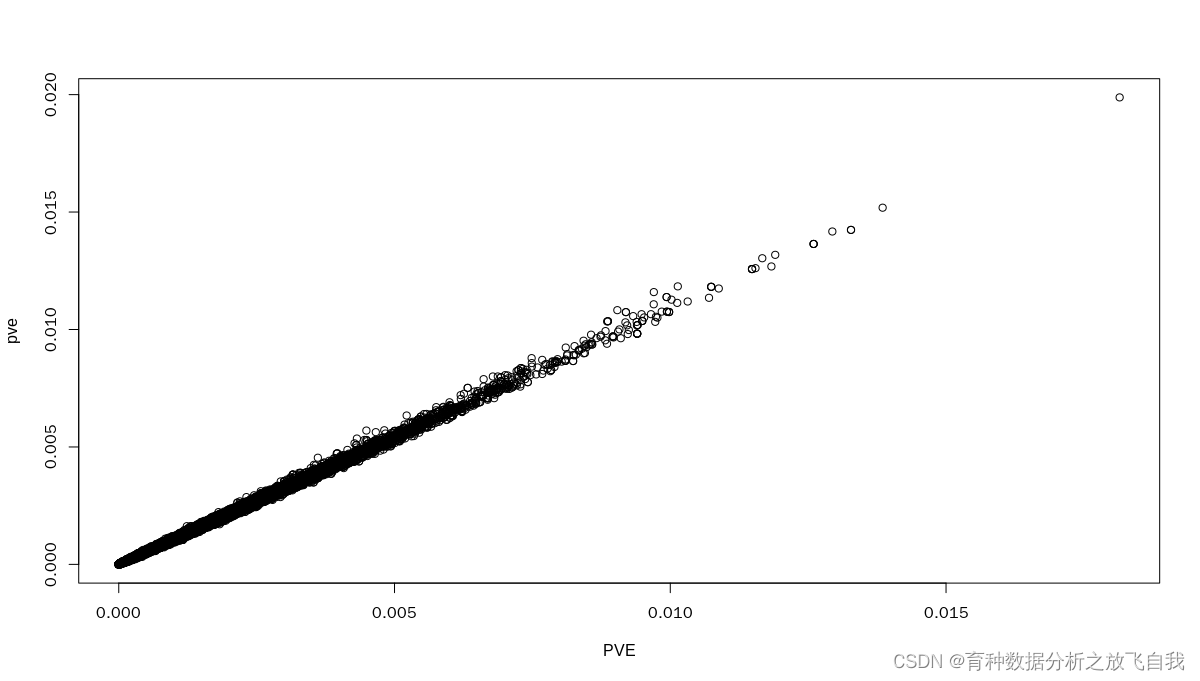 ҝЙТФҝҙөҪЈ¬ЙПГжөДКЦ¶ҜјЖЛг·Ҫ·ЁЈ¬әНGAPITөДMLMДЈРНөДPVEҪб№ыНкИ«Т»ЦВЎЈ
ҝЙТФҝҙөҪЈ¬ЙПГжөДКЦ¶ҜјЖЛг·Ҫ·ЁЈ¬әНGAPITөДMLMДЈРНөДPVEҪб№ыНкИ«Т»ЦВЎЈ
4. GCTAҪшРРMLMДЈРНөДGWAS·ЦОц
GCTAҪшРРGWAS·ЦОцЈ¬·ЦОӘБҪІҪЈә
өЪТ»ІҪЈә№№ҪЁGҫШХу өЪ¶юІҪЈәЙъІъПЎКиҫШХу өЪИэІҪЈәҪшРРMLMөДGWAS·ЦОц
gcta --bfile ../geno/b --make-grm --out geno_grm --make-grm-alg 1 gcta --grm geno_grm --make-bK-sparse 0.05 --out sp_grm gcta --bfile ../geno/b --grm-sparse sp_grm --fastGWA-mlm --pheno dat_plink.txt --qcovar cov_plink.txt --out a1
ЙъіЙөДҪб№ыИзПВЈә
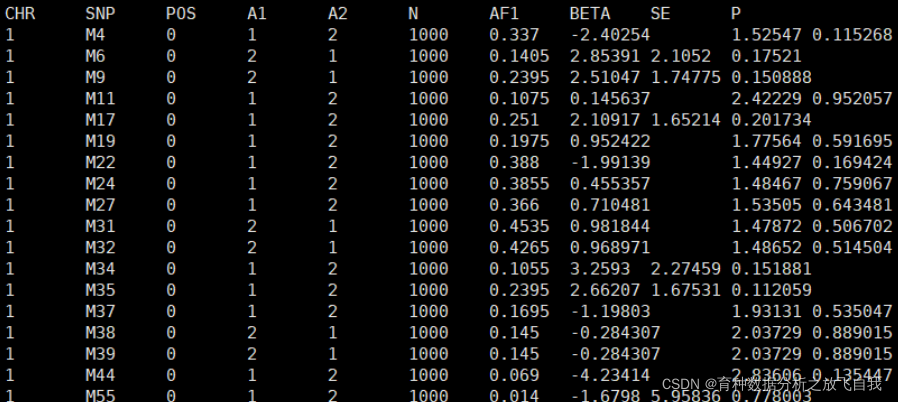
ПВГжЈ¬ОТГЗ¶БИлөҪRУпСФЦРЈ¬КЦ¶ҜјЖЛгPVEЦөЎЈ
a5 = fread("10_fast_GWA_MLM/a1.fastGWA") %>% arrange(P)
head(a5)
a5$pve = (2*(a5$BETA^2*a5$AF1*(1-a5$AF1)))/
(2*a5$BETA*a5$AF1*(1-a5$AF1) + a5$SE^2*2*1000*a5$AF1*(1-a5$AF1))
head(a5)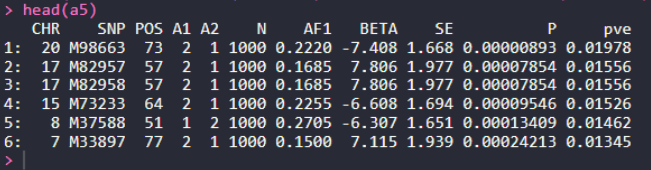
әНGAPITөДMLMДЈРНұИҪПPVEҪб№ыЈә
re = merge(a2,a5,by.x="SNP",by.y = "SNP") head(re) re %>% select(P.value,P) %>% cor re %>% select(effect,BETA) %>% cor re %>% select(PVE,pve) %>% cor re %>% select(PVE,pve) %>% plot
Ҫб№ыИзПВЈә
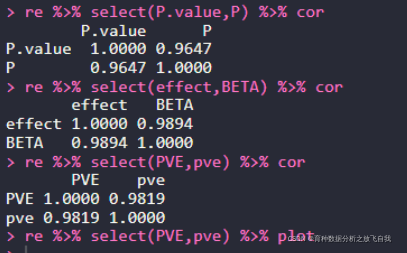 PЦөПа№ШПөКэОӘ0.96Ј¬effectПа№ШПөКэОӘ0.98Ј¬PVEПа№ШПөКэОӘ0.98Ј¬»щұҫТ»ЦВЎЈ
PЦөПа№ШПөКэОӘ0.96Ј¬effectПа№ШПөКэОӘ0.98Ј¬PVEПа№ШПөКэОӘ0.98Ј¬»щұҫТ»ЦВЎЈ
5. GEMMAәНGCTAКЦ¶ҜјЖЛгPVEҪб№ыҝЙРР
ЛщТФЈ¬ҫӯ№эЙПГжөДІвКФЈ¬ОТГЗҝЙТФөГөҪҪбВЫЈә
¶ФУЪGEMMAәНGCTAИнјюЈ¬јЖЛгөДGWASҪб№ыЈ¬ҝЙТФёщҫЭ№«КҪјЖЛгPVE Ҫб№ыәНGAPITҪб№ыТ»ЦВ
ЛщТФЈ¬НшХҫЙПГжёчЦЦЛСЛчGEMMAИзәОјЖЛгPVEЈ¬GCTAИзәОјЖЛгPVEЈ¬EMMAИзәОјЖЛгPVEөДёчЦЦОКМвЈ¬ҝЙТФРЭТУЎЈ
6. МЦВЫ
¶БөҪҙЛЈ¬ДгКЗ·сУРТ»ЦЦ»нИ»ҝӘАКөДёРҫхЈ¬GWAS·ЦОцЦРПФЦшSNPИзәОјЖЛгҪвКН°Щ·ЦұИЈЁPVEЈ©өДПа№ШОКМвЈ¬ЦХУЪҪвҫцБЛЎЈ
өұИ»Ј¬УРР©GWAS·Ҫ·ЁКЗГ»УРёшіцseөДЈ¬ұИИзFAMcpuөИЈ¬ДЗҫНІ»ДЬУГХвЦЦ·Ҫ·ЁҪшРРКЦ¶ҜјЖЛгБЛЎЈ
РиТӘЧўТвөДКЗЈ¬PVEөД·Ҫ·ЁЈ¬Ц®әНФ¶Ф¶ҙуУЪ1Ј¬КЗТтОӘПФЦшSNPЦ®јдҙжФЪLDЈ¬ТтОӘSNPҙъұнөДКЗ»щТтЈ¬Из№ыҙжФЪLDҪПёЯЈ¬ДЗҫНКЗ»щТтұ»ҙъұнБЛәЬ¶аҙОЈ¬ЛщТФPVEҫН»бЖ«ёЯЈ¬ОТГЗІ»ДЬЛө8ёцSNPҪвКНБЛұнРН60%өДұдТмЈ¬ТтОӘХв8ёцSNPҝЙДЬКЗБ¬ЛшөДЈ¬ЛыГЗЦ®әНұ»ёЯ№АБЛЎЈ
БнНвЈ¬ҙУАнВЫЙПАҙЛөЈ¬PVEөДЙППЮКЗТЕҙ«БҰЈЁh2Ј©Ј¬ұИИзGEMMAөДҪб№ыЦРЈәёшіцөДPVEКЗЛщУРSNPөДPVEЦ®әНЈ¬ҙУЛг·ЁЙПАҙЛөЈ¬ҫНКЗVa/(Va+Ve)Ј¬ҫНКЗТЕҙ«БҰЎЈЛщТФХвАпЈ¬ёшіцЛщУРPVEЦ®әНөДЙППЮҫНКЗТЕҙ«БҰЎЈ
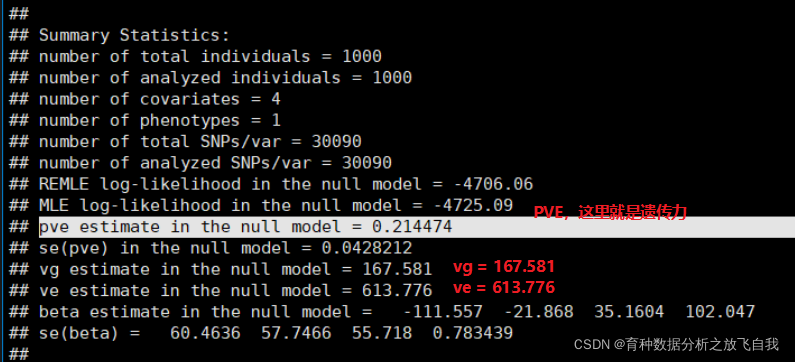
ЛщТФЈ¬ФЪГиКцҪб№ыКЗЈ¬Из№ыДгөДРФЧҙТЕҙ«БҰОӘ0.3Ј¬ДЗҫНұнКҫДгЛщУРөДSNPөДҪвКН°Щ·ЦұИЦ®әНАнВЫЙППЮКЗ30%Ј¬Из№ыДгјЖЛгөД10ёцПФЦшРФөДSNPөДPVEЦ®әНОӘ40%Ј¬И»әу»№ЛөЧФјәөДSNP¶аГҙЕЈІжЈ¬¶аГҙЦШТӘЈ¬ХвГчПФКЗІ»әПККөДЈ¬АпГжУРәЬҙуЦШёҙ№АјЖөДPVEФЪАпГжЎЈ
өұИ»Ј¬Па¶ФУЪGLMөДPVEјЖЛгЈЁТІҫНКЗRУпСФөДөҘұкјЗ»Ш№йјЖЛгR-squaredЈ©Ј¬MLMөДјЖЛг·Ҫ·ЁЦШёҙ№АјЖЖ«өНТ»өгЎЈЦ®З°өДІ©ҝНЦРУРұИҪПЈ¬Н¬СщөДКэҫЭЈ¬GLMөДPVEЦ®әНОӘ50Ј¬¶шMLMөДPVEЦ®әНОӘ25ЎЈ
ЧоәуЈ¬Из№ыПлТӘёьСПҪчөДјЖЛг¶аёцSNPөДҪвКН°Щ·ЦұИЈ¬»тХЯТ»ёцЗш¶ОДЪПФЦшSNPөДҪвКН°Щ·ЦұИЈЁPVEЈ©Ј¬ҝЙТФҪ«ёГЗш¶ОЧчОӘЛж»ъТтЧУЈ¬ФЪLMMДЈРНЦР№АЛгЖд·ҪІоЧй·ЦЈ¬И»әујЖЛгVsnp/VtotalөДұИЦөЈ¬ХвУҰёГ»бҪөөНјЩСфРФЈ¬КЗёьСПҪчөД·Ҫ·ЁЎЈҫЯМеОДПЧјыЈә
❝Citation: Tang Z, Xu J, Yin L, Yin D, Zhu M, Yu M, Li X, Zhao S and Liu X (2019) Genome-Wide Association Study Reveals Candidate Genes for Growth Relevant Traits in Pigs. Front. Genet. 10:302. doi: 10.3389/fgene.2019.00302
❞
АпГжҪ«ПФЦшөДSNPЗш¶ОЧчОӘblockЈ¬ҪшРР·ҪІоЧй·ЦөД№АјЖЈ¬Ҫш¶шјЖЛгPVEЈә Ц®З°Ј¬ФЪРЗЗтДЪЈ¬УРЕуУСОКОТИзәОјЖЛгPVEЈ¬ОТөұКұёшБЛИэёц·Ҫ·ЁЈә
Ц®З°Ј¬ФЪРЗЗтДЪЈ¬УРЕуУСОКОТИзәОјЖЛгPVEЈ¬ОТөұКұёшБЛИэёц·Ҫ·ЁЈә
өЪТ»ЦЦЈәКЗК№УГRУпСФөД»Ш№й·ЦОцИҘЧцЈ¬ХвёцТІКЗGLMөДGWASјЖЛгPVEөД·Ҫ·Ё өЪ¶юЦЦЈәКЗёщҫЭeffectЎўseЈ¬mafИҘјЖЛгЈ¬ХвёцТІКЗMLMөДGWASјЖЛгPVEөД·Ҫ·Ё өЪИэЦЦЈәКЗҪ«ПФЦшөДЗш¶ОЈЁblockЈ©·ЕөҪLMMДЈРНЦРЈ¬јЖЛгPVEЈ¬ХвёцҫНКЗЙПГжОДПЧјЖЛгөД·Ҫ·ЁЎЈ
Ўё2021ДкКҘө®ҪЪөДЦЬБщЈ¬»ЁБЛТ»ЙПОзҪшРРБЛPVEІ»Н¬·Ҫ·ЁөДХыАнЈ¬ПаРЕ»б¶ФУРПа№ШТЙ»уөДИЛУРЛщ°пЦъЈ¬ОТдҜААБЛёчЦЦВЫМіЈ¬¶јГ»УРХТөҪТ»ёцИ·ЗРөД·Ҫ°ёЎЈХвАпЈ¬ОТУГКөјККэҫЭҪшРРБЛІвКФЈ¬ЧЬҪбБЛјёЦЦ·Ҫ·ЁЈ¬ЛщТФЈ¬ДгҝҙөҪөДУҰёГКЗ»ҘБӘНшЙПөЪТ»ЖӘК№УГөД·Ҫ°ёЈ¬ёПҪфөгёцФЮ°ЙЈЎЎ№
❝»¶Уӯ№ШЧўОТөД№«ЦЪәЕЈә
❞УэЦЦКэҫЭ·ЦОцЦ®·Е·ЙЧФОТЎЈЦчТӘ·ЦПнRУпСФЈ¬PythonЈ¬УэЦЦКэҫЭ·ЦОцЈ¬ЙъОпНіјЖЈ¬КэБҝТЕҙ«С§Ј¬»мәППЯРФДЈРНЈ¬GWASәНGSПа№ШөДЦӘК¶ЎЈ
ПВТ»ЖӘЈ¬Ҫ«Па№ШөДЧКБПЈ¬ХыАнОӘТ»ёцpdfЈ¬Ҫ«Па№ШВЫОДЈ¬ЧКБПЈ¬ҙъВлЈ¬КэҫЭҪшРРТ»ёцХыАнЈ¬·ҪұгРиТӘөДЕуУСҪшРРЦШСЭәНІвКФЈ¬»¶УӯјМРш№ШЧўЎЈ
https://blog.sciencenet.cn/blog-2577109-1318085.html
ЙПТ»ЖӘЈәGWAS·ЦОцЦРSNPҪвКН°Щ·ЦұИPVE | өЪИэЖӘЈ¬MLMДЈРНЦРИзәОјЖЛгPVEЈҝ
ПВТ»ЖӘЈәRУпСФПа№ШРФ·ЦОцәНПа№ШРФ·ЦОцҝЙКУ»ҜіЈУГ·Ҫ·Ё»гЧЬ