博文
物种内circos图的绘制
|||
分析内容及数据准备(以油菜为例)
1. 基因组共线性分析:数据包括zs11.all.v0.cds(油菜基因组cds文件),zs11.v0.gff3(油菜基因组注释文件),所有工具为python版的MCscan(JCVI包)。
2. Circos图绘制:数据包括染色体核型文件karyotype.txt,需要突出展示genepair文件link.txt,染色体共线性区间文件genome.block.txt,主配置文件circos.conf。
基因组贡献分析
先将gff转成bed格式,命名为zs.bed。
python -m jcvi.formats.gff bed --type=mRNA --key=Name zs11.v0.gff3 > zs.bed
2. 然后将bed进行去重复,得到每个基因最长的转录本,产生的结果文件分别为zs.uniq.bed。
python -m jcvi.formats.bed uniq zs.bed
3. 根据zs.uniq.bed文件第4列就可以用于提取cds序列,命名为zs.cds。
seqkit grep -f <(cut -f 4 zs.uniq.bed ) zs11.all.v0.cds | seqkit seq -i > zs.cds
4. 删除第一步中的结果文件,将zs.uniq.bed文件重命名为zs.bed,,进行共线性分析,主要生成zs.zs.anchors.new文件,文件内容为genepair信息。
python -m jcvi.compara.catalog ortholog --cscore=.99 --no_strip_names zs zs
5. 将zs.zs.anchors.new文件简化为共线性block文件zs.zs.anchors.simple文件。
python -m jcvi.compara.synteny screen --minspan=30 --simple zs.zs.anchors zs.zs.anchors.new
Circos图的绘制
1. 核型文件karyotype.txt

文件分为6列,前2列保持不变。第4列为染色体实际编号,即图片上进行显示的编号,第5列和第6列表示染色体的长度,第7列实际上控制的是染色体的颜色,chr1至chr19都对应的是Circos配色体系中不同的颜色编号。
2. 需要突出展示genepair文件link.txt
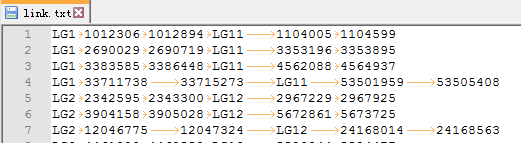
每一行代表1对genepair。共分为6列,前3列分别代表基因的染色体、起始位置和终止位置,后3列代表对应基因的染色体、起始位置和终止位置。
此文件是结合zs.zs.anchors.new文件中的genepair信息和zs.bed中的基因位置信息得来的。(不知道怎么操作的可以call我)
3. 染色体共线性区间文件genome.block.txt
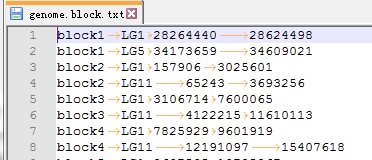
第1列代表block名称,第2-4列代表block位于的染色体、起始位置和终止位置。上下两行表示对应的block,其名称一致。
此文件是结合zs.zs.anchors.simple文件中的block信息和zs.bed中的基因位置信息得来的。(不知道怎么操作的可以call我)
4.主配置文件circos.conf
chromosomes_units=1000000 #刻度单位Mb #chromosomes_reverse=/chr[01]/ ##染染上体上的刻度逆向排列,染色体格式要是是chr1这种你就改成/chr[1]/ karyotype= karyotype.txt #染色体信息配置文件 show_tick_labels=yes show_ticks=yes spacing=10u <ticks> #设置染色体刻度 color=black format=%d multiplier=1e-6 radius=1r thickness=2p <tick> size=10p spacing=5u </tick> <tick> color=black format=%d label_offset=10p label_size=25p show_label=yes size=15p spacing=25u thickness=4p </tick> </ticks> <ideogram> #染色体绘制设置 fill=yes #是否填充颜色 label_font=default label_parallel=yes label_radius=dims(image,radius)-60p label_size=45 ##以上这四行为染色体标签 radius=0.8r #设置半径,以免基因名称过长超出显示范围 show_label=yes <spacing> default=0.005r ##两个染色体之间的间隙 </spacing> stroke_color=dgrey stroke_thickness=2p thickness=0.03r ##以上3列是对染色体轮廓设置,包括颜色,清晰度,宽度;注意颜色要在chr.info中修改 </ideogram> <links> #设置连线 bezier_radius=0r bezier_radius_purity=0.75 color=black crest=0.5 ##以上几列为links的全局的设置,包括连线的弯曲度,颜色 <link> #基因家族共线性 bezier_radius=0r bezier_radius_purity=0.75 color=set2-8-qual-1 crest=0.5 ##这几列也是连线的设置,会覆盖掉之前的全局设置 file=link.txt #输入文件,为你想在图片中的高亮文件 radius=0.98r ##连线的位置 <rules> <rule> color=green condition=var(intrachr) </rule> <rule> color=red condition=var(interchr) </rule> </rules> ##以上几列是links连线的规则,比如染色体之内(intrachr)为绿色,染色体之间(interchr)为红色 thickness=8 ##连线的厚度 z=20 #层数 </link> <link> #整个染色体上大的区块之间的共线性 bezier_radius=0r bezier_radius_purity=0.75 color=230,230,230,0.2 ##230,230,230表示灰色,0.2表示透明化(数字越小越透明) crest=0.5 ribbon=yes ##大区块之间连接为条带状,而不是连线 file= genome.block.txt #输入大区块之间共线性文件 radius=0.98r ##与前一个link的位置要保持一致 thickness=1 z=15 ##图层位置要设置得比高亮的要厚一点,不要太靠前 </link> radius=0.40r thickness=1 </links> <plots> ##显示在染色体上重点标明的基因与基因之间的关系做一个注释 <plot> color=black label_snuggle=yes #如多个文本文字距离过近,避免重叠 参考:https://www.omicsclass.com/article/678 file=text.txt #输入文件 label_font=condensed label_size=24p link_color=red link_dims=0p,2p,5p,2p,1p ##links的位置 link_thickness=2p ##厚度 r0=1r ##links起始 r1=1r+500p ##links终止 rpadding=0p ##文字边缘大小 padding=0p show_links=yes type=text </plot> type=histogram </plots> <colors> <<include etc/colors.conf>> <<include etc/brewer.conf>> #<<include etc/colors_fonts_patterns.conf>> #<<include colors.ucsc.conf>> #<<include colors.hsv.conf>> </colors> <fonts> <<include etc/fonts.conf>> </fonts> <image> <<include etc/image.conf>> </image> <<include etc/housekeeping.conf>>
5. 将以上准备文件放置一个文件夹,运行以下命令,即可出图
circos -conf config.conf

参考以下内容:
OmicsClass-circos-基因组圈图绘制软件基础绘图
TBtool软件的部分操作
https://blog.sciencenet.cn/blog-2557039-1250060.html
上一篇:关于基因家族的期刊统计及内容浅析
下一篇:批量将seq格式文件转为fasta格式文件