博文
翻译|三代测序在群体水平上的研究进展
|

对一个或多个物种的多个个体进行测序的目的是在种群水平上识别遗传变异,进一步解决进化、农业和医学等研究领域的问题。以往的包括全基因组关联研究等在内的群体水平上的研究 ,尚未完全解析人类特征和疾病背后的遗传因素。关于这种“遗传力缺失”的来源有很多猜测,如来源于结构变异和稀有变异。与常见的单核苷酸变异(SNV)相比, SV对人类基因组的变异贡献更大。迄今为止,这类群体水平上的研究主要依赖于二代短读长测序技术,读长范围从25 bp 到400 bp 不等。然而,短读长在基因组重复区域有非常大的局限性。DNA 重复序列是基因组的重要部分,是 SV 形成的热点区域,但同时也由于短读长在重复区域比对不准确而阻碍了 SV 的鉴定。然而即使在基因组的非重复区段,如插入(特别是对比短读长更长的等位变异)或其他修饰(例如,甲基化)等这样的变异也会被单纯依赖于短读长的方法所忽略。
在结构变异鉴定上,三代长读长测序优于二代测序和其他方法(例如芯片)。瓶中基因组联盟和人类基因组结构变异联盟的研究结果表明,长读长可发现相当多的之前短读长未发现的变异。最近冰岛和中国人群的长读长测序研究发现了以前未被发现的与身高、胆固醇水平和贫血关联的变异。分析26个玉米基因组发现,SV主要与玉米的抗病性状而不是农艺性状相关联。
测序技术和生物信息学的不断进步为群体水平的长读长测序研究铺平了道路。推动该领域创新的两个主要竞争对手是太平洋生物科学公司(PacBio)和牛津纳米孔技术公司(Oxford Nanopore Technologies)。PacBio公司的Sequel II 系统可产生长度长(15-20 kb)且精确度高的HiFi read。ONT 公司的PromethION 平台可产生更长的read (高达4 Mbp) ,且通量高、成本低,但产生的read准确度低于 PacBio公司的Sequel II 系统。最近一项研究发现,这两个平台在SV鉴定方面旗鼓相当。在过去的两年中,多项研究应用三代测序来回答多种生物学问题。目前三代测序主要在人类上应用较为广泛,三代测序在作物、果蝇和家禽等群体基因组学研究上也有少量应用,在宏基因组学研究中也发挥了越来越大的作用。
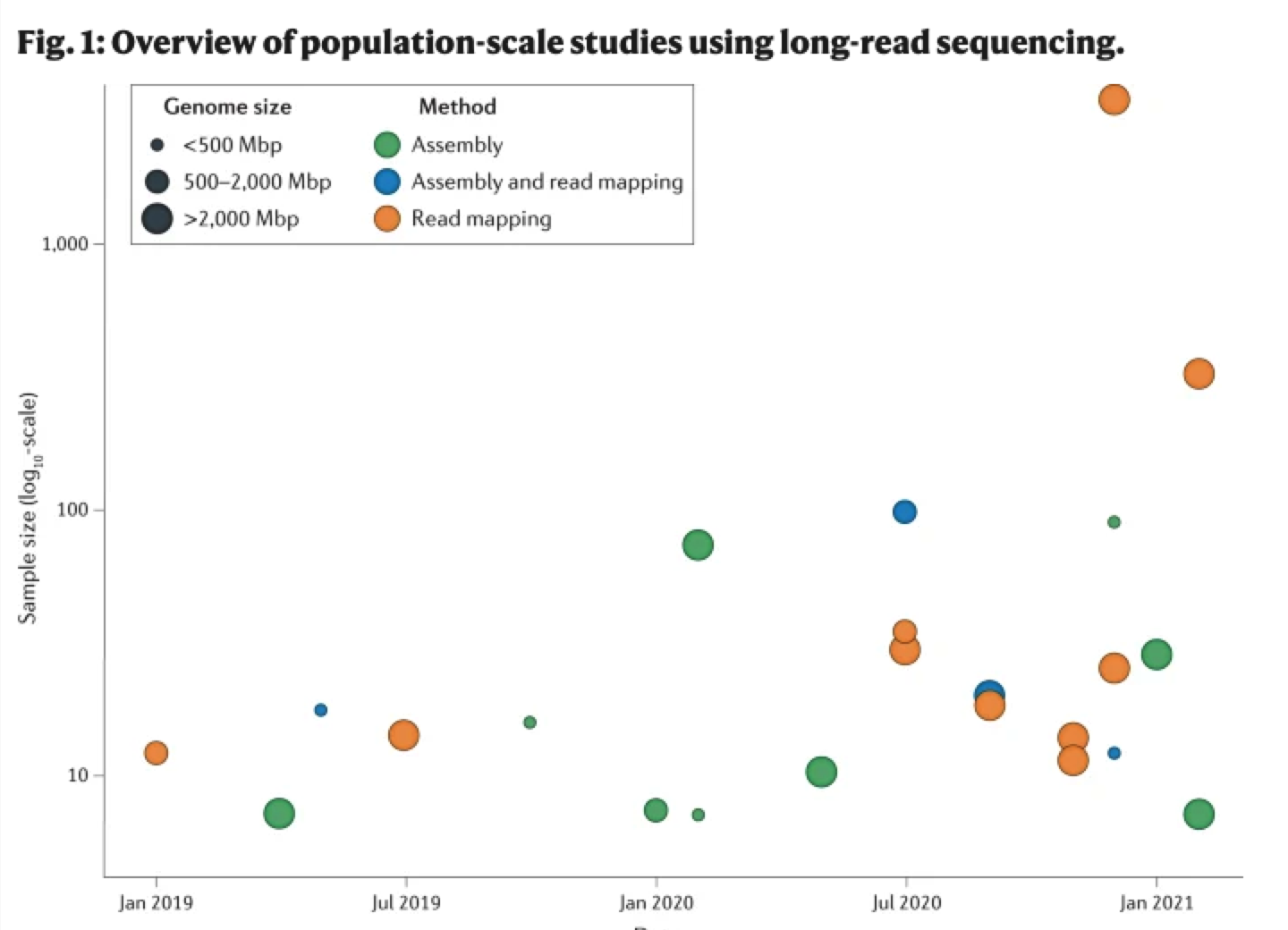
图一:群体水平三代测序研究总览
Project strategies
一个群体中要选择多少个体进行测序,要具体问题具体分析,视具体的科学问题而定。虽然可采用低至5个个体估计遗传分化或祖先种群大小 ,但是鉴定种群的罕见变异需要的样本数量通常要高几个数量级。但是,无论采用何种方法,群体的协变量控制以及元数据的完整性都是至关重要的。
一个大规模的群体测序项目,根据预算需求,通常有几种策略可选择。根据项目预算、研究目标等,本文主要讨论了三个具有不同遗传变异检测分辨率的策略。在几乎所有的测序技术中,测序成本一直在降低。为了能够比较这三种策略,我们做了一些近似的估计以及简单化处理。虽然我们假设二倍体基因组的大小与人类单倍体基因组相似,但我们注意到,对于倍性较高的基因组(例如一些六倍体植物) ,整体覆盖度必须达到生物体的倍性(即同源染色体数目)的最低需求。此外,我们假设样本大小为2500个个体,这类似于千人基因组计划。在2021年初编写本报告时,生成长读长测序数据的最便宜选择是 ONT公司的 PromethION 平台,每个流动槽的获得约为100-150 Gbp的数据,价格在650美元至2100美元之间,具体取决于购买多个流动槽时所获得的折扣。值得注意的是,PacBio HiFi 的read有足够的长度和高精确度,虽然没有正式评估,但对这项技术来说有理由相信较低的基因组测序覆盖度就能达到要求。然而,在撰写本文时,成本仍然比 ONT PromethION 平台高,PacBio一个SMRT cell的成本约为1300美元,产生约500 Gbp (连续长读)或约30 Gbp (HiFi)的数据。
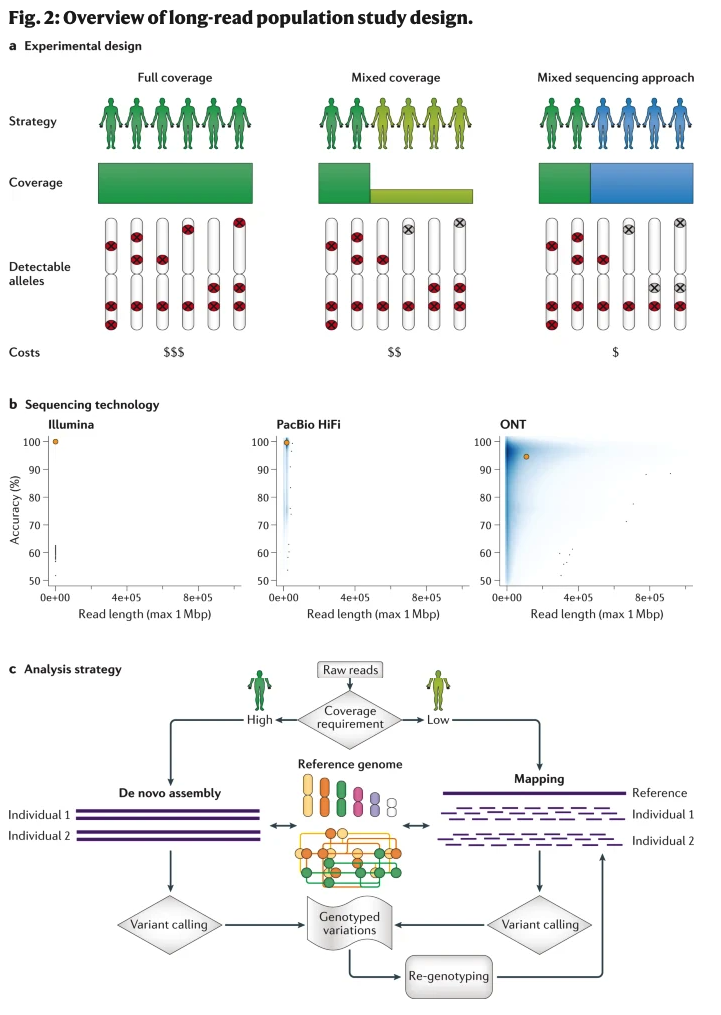
图二 三种不同的测序策略
A full coverage approach
对群体中的每一个个体进行中覆盖度或高覆盖度测序是一种分辨率最高的检测方法,但也是三种方法中最昂贵的方法。每个样本的测序覆盖度取决于项目计划和目标,是进行从头组装(需要覆盖度大于40倍)还是采用与参考基因组比对的方法(需要覆盖度大于12倍)。这种策略的优点是它的全面性、研究设计的简单性和相对简单的计算工作流程。样本的覆盖度相似,系统误差或批次效应等也较容易消除,而且每个样本中罕见的变异也可以检测出来。以20倍覆盖度测序所有2,500个人的样本约产生150 Tbp 的测序数据。
A mixed coverage approach
“高低覆盖度混合”方法就是对群体的每个亚群中具有代表性的个体进行高覆盖度(如30倍)测序,剩下的个体进行低覆盖度测序(如> 5倍)。虽然这种方法一般比前一种方法便宜,但仍然具有较高的总体分辨率,因此特别适合于样本数量多或预算有限的研究。然而,仍然存在一些分析方面的挑战,特别是整合多个样本时还能保证基因分型的高准确性,实际情况只能会更加复杂。此外,这种混合覆盖度测序方法肯定会偏向于常见等位基因,因此可能会遗漏许多稀有等位基因,特别是如果一个位点是杂合的,次要等位基因可能会被掩盖。假设在第二个方法中,200个人以30倍覆盖度测序,其余人以8倍覆盖度测序,这种方法就只需要73 Tbp 的数据,因此只需要一半的费用。
A mixed sequencing approach
"长短读长混合测序"方法对少数样品(如所有样品的10-20%)进行三代测序,然后对剩余样品进行二代测序。这种方法的基本原理类似于在混合覆盖度测序方法中先确定一小部分样本(或是随机或根据多样性、种族或表型等) 进行高覆盖度测序。在大豆中利用二代测序技术获得的变异进行系统进化分析,筛选出一组具有代表性的大豆材料进行三代测序并进行从头组装。其他一些研究则使用 了SVCollector这款软件选择样本进行三代测序。一旦用三代测序技术对队列子集进行测序,获得SV后以此为参考,就可以对二代测序数据集进行基因分型 (如大片段插入)。通过这种方法,已鉴定变异的等位基因频率能够可靠的获得,尽管偏向于利用三代测序获得的变异,但这意味着其他样本中的罕见变异可能被遗漏。通过二代测序直接对所有类型的 SV 进行基因分型是不可能的,特别是在重复区域,但是能够基于二代测序获得的 SNV 基因型进行基因型填充。这个策略已经被应用于人类的研究中用以发现新的eQTL和适应性进化特征。如果这一方法在已有二代测序数据的基础之上,那么这种方法可能是最经济的,因为只需对2500个人中的200个人进行30倍覆盖度测序,而这只需要18 Tbp 的数据。
Sequencing logistics
文中还对PacBio和ONT平台在DNA准备,设备维护,数据的保存等进行了分析。这部分可能我们大多数人接触不到,现在网上也有很多资料介绍三代测序的原理,这里就不在详细阐述了。
Analytical considerations
可以说,群体水平上的研究的主要挑战是开发一个可扩展以及稳健的分析流程。许多最近的综述讨论了在单个样本中的方法,表2列出了在三代测序项目中常用的计算工具。值得注意的是,在这个发展非常迅速的基因组学领域,还没有公认的分析流程或标准,新的工具不断开发出来,而已有的工具很快就会过时。这里没有讨论二代和三代整合在一起的分析,但不能否认二代测序数据在校正三代数据中所起的作用。
表2 三代测序中的分析工具汇总
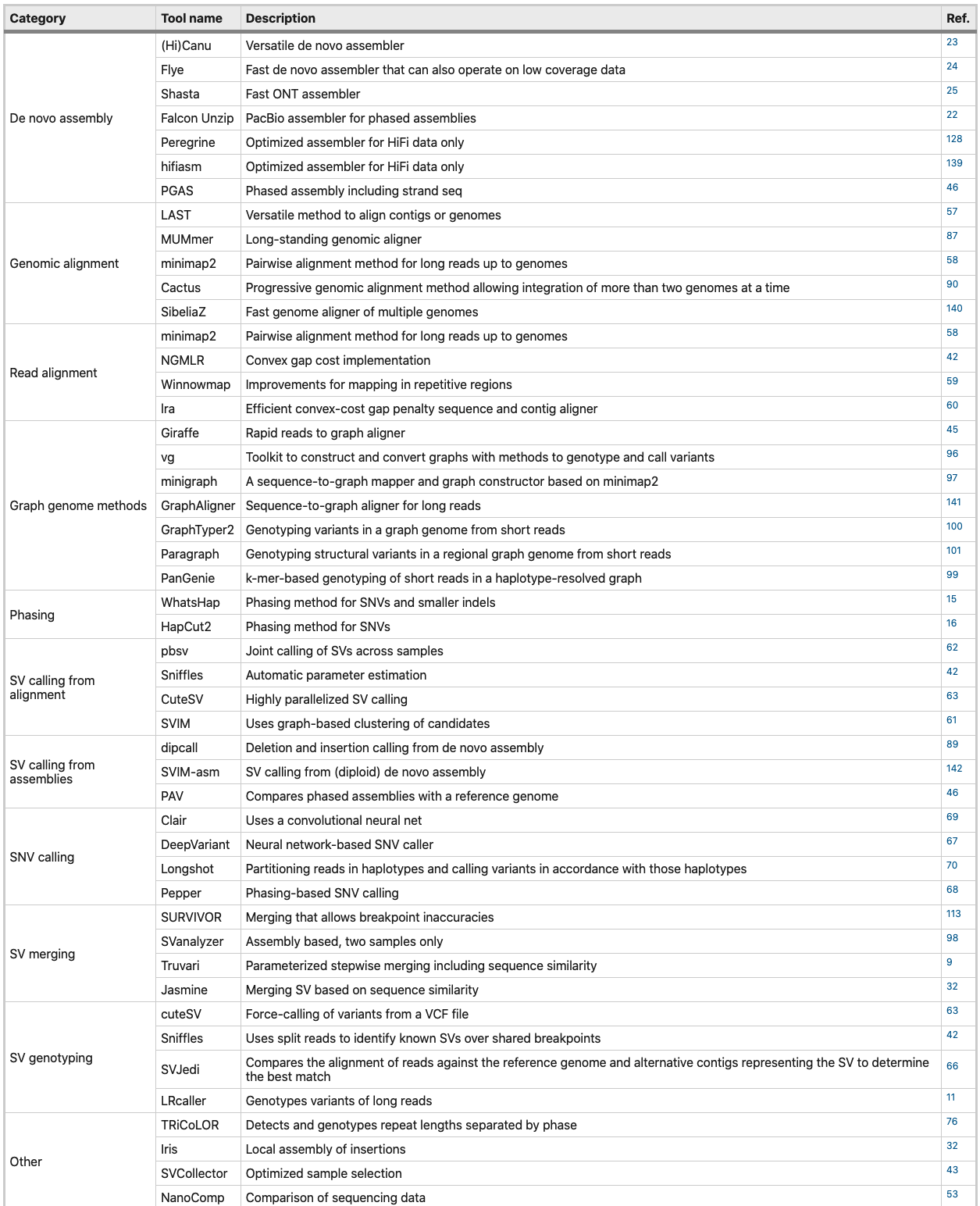
对群体水平上的项目而言,分析工具的选择往往在结果的准确度和计算效率之间平衡。在进行下游分析之前,对直接影响组装性能、 SV 检测和read定相等方面的实验因素如 DNA 片段长度和测序产率进行质量控制至关重要。为此,已经开发了多种工具。项目期间测序设备试剂等因素的变化可能会导致批次效应,从而影响结果。因此,要合理安排样品,尽量避免批次效应以及其他无关变量的影响。有两种主要的下游分析策略可供选择: 将单个样本测序数据比对到参考基因组、先从头组装基因组再进行基因组水平的比对。这两种方法在计算和测序覆盖度要求上有很大的不同,而这又在很大程度上取决于基因组的大小和复杂度。对于这两种方法,目标都是对所有样本应用相同的方法以及软件版本。结果需要以一致的方式生成,使用正确的版本控制和可复制的分析流程,以避免分析中引入额外的错误。在下面的章节中,我们讨论与参考基因组比对、从头组装以及图形基因组的方法。
Read alignment-based analysis
与参考基因组比对的方法通常是群体水平研究的选择方法,一半以上的群体基因组学研究使用了该方法,因为它们有助于将所有样本与一个共同的坐标系(即参考基因组)进行比较。此外,与从头组装相比,该方法往往对计算机的配置和测序覆盖度的要求较低。基于比对的方法依赖于测序的read与参考基因组的匹配,其整体准确性将影响数据分析。如果参考基因组不完整、不正确、支离破碎或与目标样本遗传差异过大,就会导致下游分析的偏差。
近年来,三代测序数据的分析软件层出不穷,而且比对速度越来越快。NGMLR 和 LAST 软件加快了比对过程,提高了比对的精确度。minimap2比其他比对软件快得多,而且结果也类似,因此它是目前最流行的,被广泛接受的三代测序数据比对软件。最近两个值得注意的软件是 Winnowmap,它改进了复杂区域的比对,特别是重复区域的比对 ;另外一个是 lra,它改进了有 SV时的比对。
选择什么样的工具检测遗传变异同样非常重要。目前有几个可用的工具可以用来鉴定SV,如 Sniffles、 SVIM、 PBHoney、 CuteSV 和 pbsv 。剩下的挑战之一是如何准确判断 SV 断点,尤其是在群体水平以及复杂区域。虽然最近开发的工具削弱了 SV 分型对高测序覆盖度的需求,但绝不是尽善尽美的,并不说要完全替代高覆盖度的测序,还是要具体问题具体分析。
基于二代测序开发的鉴定 SNV 和插入缺失(InDel)的方法应用到三代测序上并不理想。过去几年中,已经开发了多种策略,用于改进每种三代测序检测小插入缺失的准确度。目前的方法包括 DeepVariant Pepper、 Clair 和 LongShot (明确要求等位基因与单倍型结构一致) ,其准确性高于使用illumina数据的方法。与 ONT 公司相比,PacBio HiFi 模式在小InDel方法的表现可与 Illumina 平台媲美。
串联重复变化频繁,对这种区域分型挑战很大。这些短串联重复序列和小型重复序列与致病变异有关,因此准确的等位基因分型是至关重要的。一些专用工具也已经开发出来了,如tandem-genotypes 和 TRiCoLOR。类似的挑战仍然存在于其他类型的重复。例如,LPA 位点由8个编码 kringle IV 结构域的串联重复单位组成,在人类基因组中重复5-10次,因此很难评估。
到目前为止,大多数参考基因组都不是单体型的,即父母两套染色体合并在了一起。基因组组装定相,能够获得每条染色体序列,从而能更准确地代表基因组。人类端粒到端粒联盟的目标是产生第一个完整的人类染色体组装,并且已经完成了8号染色体和 x 染色体的组装。在另一个例子中,在乌鸦群体遗传学研究中,一个单体型基因组就用作比对的参考。
Population-scale de novo assemblies
基于二代测序的参考基因组,序列上不完整,高度碎片化。此外,数百Mb的种群和个体特异性序列并不存在于人类参考基因组中。这些丢失的序列往往是重复序列,但也包括编码序列。因此,总有一小部分序列不能比对到参考基因组上或者比对到错误的地方,从而导致错误的分型。因此,先从头组装基因组再进行基因组水平的比较是可取的。
随着三代测序数据技术的进步以及成本的下降,更快和更准确的基因组组装工具也成群结队的出现 。获得完整的没有缺口的单体型基因组是基因组组装中的极致追求,一些新工具也迅速发展起来,这些工具通常需要权衡计算能力、序列连续性、完整性或正确性等关键参数。与基于序列比对的方法相比,从头组装的方法往往更灵敏,更适合于重建高度多样化的基因组区域,但有时候对高度重复序列也无能为力。对于这些重复区域,已经开发出专门的算法,如利用 SNV 区分多个重复序列,从而可以恢复这些重复基因。从头组装方法依赖高覆盖度测序和更多的计算需求,所以在对群体水平上的基因组进行从头组装挑战非常大。
单基因组项目可以有很多时间用来测试多个参数或不同的方法来优化从头组装,但这在群体水平是不现实的。对大量的样本来说,从头组装方法通常需要更多的计算资源。虽然大型云计算基础设施可以满足这一需求,但计算成本将大幅上升。所以,迫切需要发展对计算要需求较低的组装工具。
另一个重要的考虑因素是下游分析的可扩展性。虽然基因组组装过程已经需要大量的计算资源,但下游分析对计算资源的需求会随着个体数量的增加而增加。为了可以系统比较基因组之间的变异,通常将从头组装的基因组与选定的某个参考基因组进行比较,从而产生一个标准的VCF格式文件。目前,一些工具如 MUMmer,Assemblytics,miniap2,dipcall和 SVIM-asm可以进行2个基因组的成对比较。然而,在多个基因组比较的项目中,如果都与同一个参考基因组进行比较,这就与基于二代测序的方法具有相同的偏差。例如,参考基因组中没有的序列就难以在所有样本中进行比较(图3a)。这个问题可能会因参考基因组gap的存在而更严重。虽然基因组水平的比对费时费力,但这种付出是值得的,与基于短read比对的方法相比,可以获得更多更复杂的SV(图3b)。最全面的选择可能是两两比较的方法(图3a) ,但比较的次数会按二次方增加,这意味着在有100个样本的情况下,就有4950次比较。显然,这种方法目前对大多数项目来说是不可行的,必须制定替代方法。最近,Cactus工具的发展,使得多个基因组的比较成为可能。然而,到目前为止,这个工具主要是跨物种比对,而不是在物种内不同个体之间比较。
.png")
图三 多个基因组之间比对面临的问题
从头组装方法面临的另一个更大的挑战是获得正确倍性的基因组。许多生物具有二倍体基因组(例如,人类和许多动物) ,而一些作物的基因组倍性更高。优化二倍体单体型-敏感的从头组装工具可用于重建这两种单体型。这种重建对于恢复所有的杂合变异至关重要,否则两个不同的单倍型会被压缩成一个人为的、不正确的染色体。然而,单体型分辨率的从头组装方法通常需要更高的覆盖度和计算资源。杂合变异和纯合变异的正确基因分型对随后的群体遗传分析至关重要。最近的一个解决方案是首先创建一个不分单体型的组装,然后将变异和reads分成单倍型,最后将contig定相。即使获得的单体型基因组具有良好的精确度,对高度重复区域进行SV鉴定可能也会无能为力。例如,具有高重复性结构的人类 LPA、SMN1 和SMN2 基因位点在比对时总会碰到问题。因此,主要的挑战可能转移到基因组比对和不同基因组之间差异如何准确表示上。
Graph genome methods
read比对和从头组装方法都可能存在系统性问题,如复杂的结构变异、参考基因组中缺失的序列、重复区域的变异和高度多态位点。线性参考基因组只代表一个等位基因,没有整合群体中的多态性位点以及复杂区间。泛基因组方法是将同一物种内多个个体的基因组整合在一起的方法,更能表示基因组的多样性。泛基因组构建所需的结构变异也是通过前期获得的高质量的个体基因组之间的比较获得的。在read比对时考虑已知的变异,可以减少比对偏差。因此,这不依赖于单一的参考基因组。在这方面已经有一些工具开发出来,如vg、 minigraph、 Seven Bridges Graph Genome Pipeline、 DRAGEN Graph Mapper 和 PanGenie等。图形基因组方法的另一个好处是,它们能够更正确地表示嵌套变异,例如插入序列中的小变异。
图形基因组一个主要好处是可以使用二代测序数据对SV分型。目前一些工具如GraphTyper2、 Paragraph 和 vg 包中的工具,专门用于二代测序数据与图形基因组的比对。一个样本中的 SNV、小InDel或 SV 可通过图形基因组的特定路径进行基因分型。图形基因分型方法能够发现原先基于二代测序数据鉴定SV方法中所不能鉴定的新的SV变异。新发现的结构变异可以整合到泛基因组中,进一步提高分型的准确性。

通过这种基于图形的方法,在群体研究中可以避免一些顾虑,即是使用现有参考基因组还是从头组装一个新的参考基因组进行比对。因为在下一步的分析中,所有序列必须与泛基因组进行比较,以识别变异、注释和统计评估。然而,这些方法在实践中没有使用线性参考基因组那么简单,而且还不完全成熟,在具体实现和数据格式等方面还没有形成一个共识。虽然图形基因组方法是避免基因组多样性偏差的好方法,但目前尚不清楚这些方法是否会成为临床诊断应用的主流。
Variant validation and genotyping
为了确定鉴定出来的变异是真实存在的,通过多种手段进行验证就变得非常重要。理想情况下,采用不同方法之间鉴定的变异进行互相验证。传统的PCR 方法不适用于高重复区域 SV的验证,而光学图谱可能更适合。目前通过人眼查看和判断可以说是一种非常准确的验证方法,但是对于含有几百个个体的群体来说肯定是不可行的。目前一个半自动化的命令集SV-plaudit (Samplot) 已经开发出来,可以高效的进行成千上万SV的验证。
对于 SV来说,情况要复杂得多,因为在不同样本之间获得一致的变异并不简单。首要的方法之一是当两个SV有50%重叠时就进行合并。虽然这对于大的拷贝数变化事件非常有效,但对于具有更多局部断点的较小的SV (例如,50 bp 到1 kb)可能有一些限制。另一种方法是要求每个个体的断点近似一致,以确定两个样本中的变异确实是同源的。在某些情况下,如两个插入是同源的,但是它们的序列有轻微的偏差,基于断点的方法可能过于保守,并且已经有一些工具试图解决这个问题(例如 Truvari、 SVanalyzer 和 Jasmine)。然而,目前还没有通用的阈值标准。因此,这些方法依赖于断点距离和序列相似度。缺失可以说是结构变异最直接的变异类型,但即使是这种看似简单的 SV 鉴定杂合子却不容易。像 Sniffles 和 SVJedi 这样的工具能够根据候选 VCF 文件对 SV 进行基因分型,这是在基于三代测序read比对发现 SV 的第一步。
提高 SV 基因型准确性的另一个非常有效的方法是利用遗传距离信息。在这种方法中,基本的群体遗传假设被用来减少基因型 SVs 的假阳性数量。经过足够数量的世代(4Ne,Ne 是有效种群规模)后,变异可能已经完全固定,在同一个亚群(即无限位点模型)内不应再发生变异。在不同谱系中显示多态基因型的任何变异都被排除在外。虽然这种方法忽略了某些类型的 SV 具有更高的突变率,因此确实具有重复突变的潜力,但它为获得可靠的 SV 基因分型做了有益的尝试。这种方法最近已成功地应用于鸦类和寒鸦群体基因组学研究中。
Prediction of functional impact
虽然使用 Ensembl VEP 等工具对突变影响蛋白质功能的分类已经相对成熟,但要判断 SV 对附近基因表达的影响就不那么容易了。这主要是因为还不清楚 SV 的长度如何影响周围的基因组区域,而且通常很难获得稳定的 SV 等位基因频率。对于功能注释和致病性预测,使用联合线性模型、监督式学习和现有数据库的方法已经被开发出来,并且一些例子表明 SV确实与重要的表型有关。
Conclusions
正在进行的重大技术改进将为三代测序应用于群体水平的基因组学项目铺平了道路,并表明这种测序方法将长期存在。对群体水平的三代测序分析仍然具有挑战性,目前最可行的方法是基于read比对的方法。尽管如此,我们预计这将为改变单体型敏感的基因组或图形基因组。这一发展将对该领域产生深远的影响,并有希望提高变异的代表性和复杂性,但需要系统的从线性参考基因组到更复杂基因组的转变。
PacBio 和 ONT 领导了长读长测序技术的发展。然而,其他公司(例如 Base4、 Quantapore 和Omniome)正在开发新的长读长测序技术,其可行性仍有待在未来几年内进行评估。虽然这里没有讨论,但 DNA 提取的改进、保存和文库准备工作也加速了长读长测序在群体水平上的应用。近年来最大的成就之一是生成4 Mb 或更长的read。将来一旦常规的测序读长也能接近染色体长度,基因组组装似乎过时了; 然而,这样的read能否让从头组装方法替代基于比对的方法仍有待观察。
Future directions
未来群体水平的长读长测序为多种类型的组学研究提供了许多机会。例如,PacBio 和 ONT 平台都能够同时检测核苷酸序列和 DNA 修饰,比如5-甲基胞嘧啶。这种修饰的鉴定对表观遗传学和 DNA 损伤分析具有前所未有的意义。一些研究表明,纳米孔测序可以成为检测甲基化模式的金标准,但现在还缺乏相关的统计方法。核苷酸修饰的检测进一步给染色质可及性分析和复制叉检测等方面的分析带来了新的机会。
与基于 DNA的群体水平的测序相辅相成的是,mRNA 和cDNA 的长读长测序也可以用来鉴定可变剪切的多样性。为了研究已知的和新的异构体,人们已经开发了许多分析工具,但这一领域还远未成熟。长读长测序方法也已经扩展到单细胞转录组学。虽然这些应用可能会带来生物学上的奇妙见解,但是对于群体水平上的研究的影响还不清楚。
随着长读长测序技术以及计算分析工具的改进,以前需要数周到数月才能完成的分析现在可以在一天到一周内就能完成,而且成本更低。然而,一些挑战仍然存在,例如,如何表示嵌套和高度复杂的变异。最近的进展,如泛基因组,有可能解决这一挑战。此外,基于图形的泛基因组的使用确实可以改善分析本身,因为它包括了不同的等位基因,所以克服了单个参考基因组的偏差。另一个相关的计算挑战是对复杂等位基因进行准确、快速的分型。在这方面,图形基因组有着天然的优势。
尽管长读长测序方面取得了重大进展,但仍有若干挑战有待解决。比如,三代测序的准确性。另外,评估复杂区域的变异仍然存在困难,例如片段复制、核糖体 DNA 串联重复、端粒或着丝粒。但令人兴奋的是,获得0缺口的基因组已成为现实。
https://blog.sciencenet.cn/blog-1094241-1293869.html
上一篇:翻译:植物泛基因组综述(Plant pan-genomes are the new reference)
下一篇:python httpx 异步爬虫