博文
活性氧诱导核毒性应激反应导致代谢性衰老的基础
|
ROS 诱导的核糖体损伤是 ZAKα 介导的肥胖和衰老代谢下降的基础
核糖体氧化损伤可能是细胞水平老化的关键体现。 ZAKα是一种MAPK,在核糖体氧化损伤中发挥关键作用。本研究发现, ZAKα是核糖体氧化损伤的分子调节基础。阻断该过程能延缓肥胖和衰老相关的代谢异常。由于慢性代谢损伤涉及到脂肪肝和代谢综合征,这一核糖体损伤的分子基础,为解决慢性病提供了一种新的研究策略。
在食物匮乏的条件下,代谢的灵活性对于人类在食物不容易获得时优化储存和获取能量具有重要意义。在现代社会中,不断获得高热量食物已成为一种负担,导致肥胖和代谢疾病的增加。活性氧被认为会导致与肥胖和衰老相关的代谢失调,但其潜在过程尚不清楚。Snieckute 等确定了蛋白质ZAKα可将活性氧与核糖体功能变化联系起来的机制,有助于在多个模式动物物种中观察到的代谢变化。
结构化摘要
介绍
当核糖体在 mRNA 翻译过程中停滞和/或碰撞时,核糖体监视机制被激活。这些途径表明存在核毒性应激,并协调非生产性核糖体的解析。核糖体结合和应激感应 MAP3 激酶 ZAKα 位于核毒性应激反应 (RSR) 的纽带,最终通过应激相关 MAP 激酶 p38 和 JNK 激活和信号传导。RSR 通路是非必需的,对发生翻译问题的体内条件以及 RSR 在生理上很重要的背景知之甚少。
理由
活性氧 (ROS) 激活 p38 和 JNK 并损害核糖体翻译。我们认为这些反应可能是偶联的,并且与一系列生理和病理生物学转变相关的 ROS 是体内 RSR 激活的来源。ZAK 敲除 (KO) 小鼠表现出轻度代谢表型,瘦弱且肥胖减少,表明 RSR 参与代谢调节。此外,p38 和 JNK 参与代谢转化,例如肥胖症中胰岛素抵抗和肝脂肪变性的出现。在肥胖和代谢调节的背景下,这些激酶的激活是由难以捉摸的代谢应激信号驱动的,这些信号可能由21种已知的p38和JNK定向的MAP3激酶中的任何一种介导。因此,我们试图使用细胞、斑马鱼和小鼠模型研究 ROS、RSR 信号传导和代谢调节之间的潜在联系。
结果
我们证实 ROS 激活细胞模型中的 RSR,与核糖体的停滞和碰撞有关。体外翻译反应强调,这些效应主要是由对可溶性翻译因子(包括转移RNA)的影响介导的。当暴露于ROS的病理性爆发时,缺乏zakα基因的发育斑马鱼幼虫受到强有力的保护,防止早期致死,这可能是由于预防RSR诱导的程序性细胞死亡。ZAK KO小鼠喂食高脂肪高糖(HFHS)饮食,这与啮齿动物的快速体重增加和ROS驱动的病理有关,可以防止代谢适应不良的早期表现,包括血糖不耐受和肝脂肪变性。这些表型伴随着ZAK KO小鼠肝脏中失调的RSR信号转导。在HFHS喂养的小鼠中,高度翻译的肝脏mRNA显示出核糖体的停滞和排队增加,表明存在已知的ZAKα激活结构。最后,雄性小鼠的衰老还与ROS产生增加和代谢恶化有关,表现为血糖不耐受受损,随机肝脂肪变性和棕色脂肪组织变白。ZAK KO小鼠受到保护,免受代谢衰老的所有这些标志的影响。
结论
我们的研究揭示了 ROS 是平移畸变和 RSR 激活的生理相关来源。此外,受损的核糖体至少构成代谢应激信号的一部分,这些信号会导致肥胖和衰老中不必要的代谢适应不良。Zak基因缺失在小鼠中对这些反应提供了强有力的保护,因此有必要研究ZAKα激酶作为代谢性疾病(如非酒精性脂肪性肝炎,高血压和血脂异常)的潜在药物靶标。需要更多的工作来揭示RSR信号转导对上述表型的组织特异性贡献,并确定核毒性应激的其他生理相关来源。
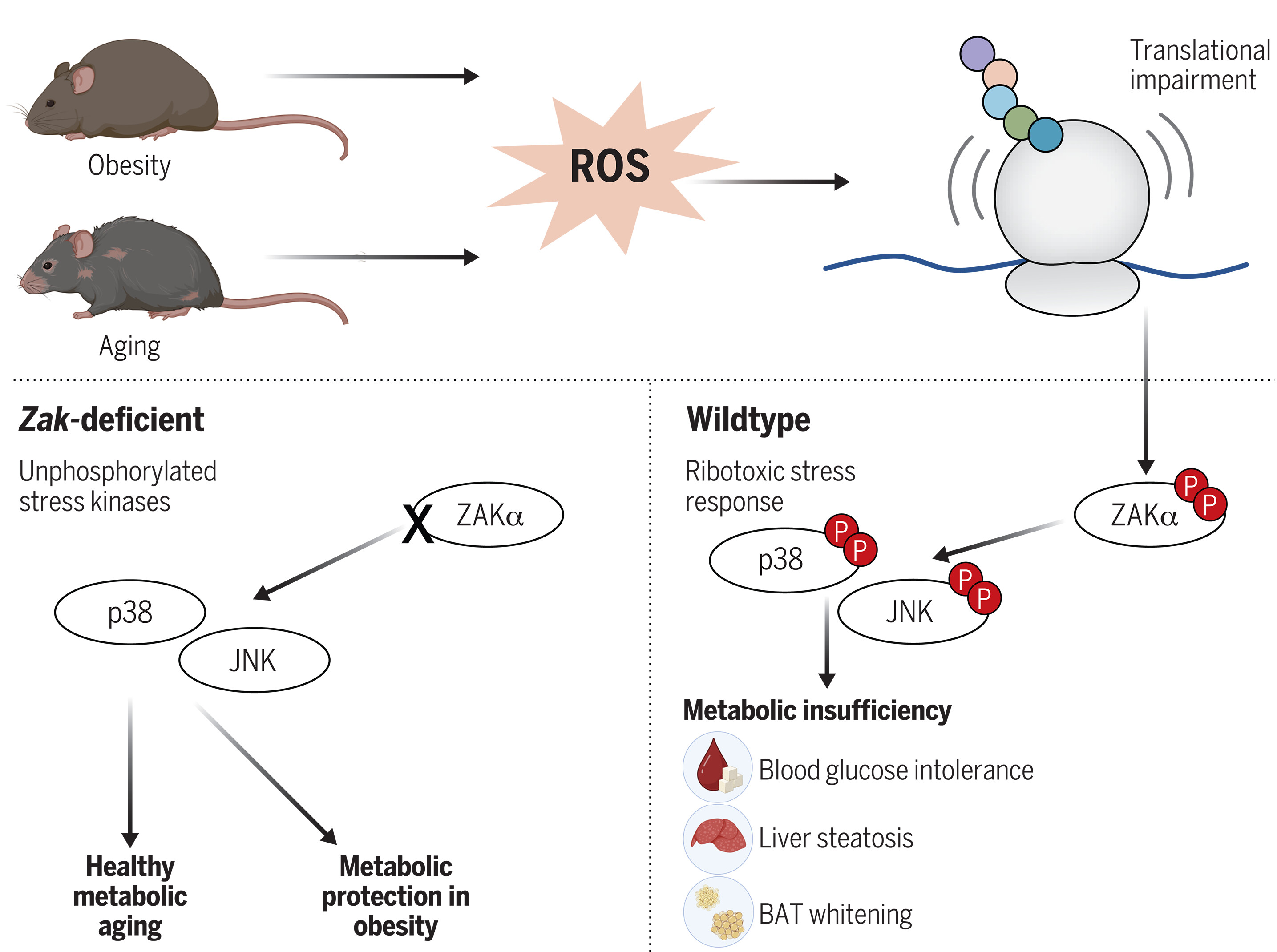
核毒性应激反应驱动肥胖和衰老的代谢适应不良。
ROS 是应激激酶 ZAKα、p38 和 JNK 的翻译损伤和激活的生理相关来源。在肥胖和衰老的小鼠中,这种 RSR 驱动代谢适应不良,表现为血糖不耐受、肝脂肪变性和棕色脂肪组织 (BAT) 变白。
核毒性应激反应 (RSR) 是一种信号通路,其中 p38 和 c-Jun N 末端激酶 (JNK) 激活的丝裂原活化蛋白激酶激酶激酶 (MAP3K) ZAKα 感知核糖体的停滞和/或碰撞。在这里,我们表明活性氧 (ROS) 生成剂触发核糖体损伤和 ZAKα 激活。相反,缺乏 ZAKα 的斑马鱼幼虫免受 ROS 诱导的病理学的影响。喂食产生ROS的饮食的小鼠肝脏在核糖体伸长动力学中表现出ZAKα激活的变化。突出RSR在代谢调节中的作用,ZAK敲除小鼠免受高脂肪高糖(HFHS)饮食诱导的血糖不耐受和肝脂肪变性的影响。最后,ZAK消融术减缓了动物发展代谢衰老的标志。我们的工作强调 ROS 诱导的核糖体损伤是 ZAKα 的生理激活信号,是肥胖和衰老代谢适应的基础。
高度的代谢灵活性允许在资源稀缺时优化能量的释放和利用,并在资源丰富时有效地储存能量。 在当代社会,持续获得富含卡路里的食物导致了全球肥胖流行病。在这种情况下,动态代谢调节的有益机制会对体内平衡产生负面影响。需要更深入地了解潜在的信号转导通路,以指导肥胖和相关代谢疾病的新治疗原则的开发。这些疾病包括 2 型糖尿病、非酒精性脂肪性肝炎 (NASH)、高血压和血脂异常。早期肥胖相关代谢功能障碍的特征包括胰岛素抵抗、胰腺β细胞功能不全、肝脏脂质积聚(脂肪变性)和脂肪组织肥大。在衰老过程中也会发生类似的变化,这表明代谢改变的潜在机制是相关的。代谢失调的一个潜在驱动因素是活性氧(ROS),其产生在肥胖和衰老中增加。 升高的 ROS 有可能扰乱细胞的氧化还原平衡并破坏蛋白质、DNA 和 RNA 等大分子。生理范围内的 ROS 是具有有益功能的稳态的先决条件信号转导分子。目前尚不清楚升高的ROS如何在生物体尺度上扰乱代谢功能,其潜在机制可能包括对大分子的不加选择的氧化损伤和代谢信号通路的调节。
应激激活的丝裂原活化蛋白激酶 p38 和 c-Jun N 末端激酶 (JNK) 被多种细胞应激因子激活,例如 ROS、紫外线 (UV) 光、热应激和机械扰动。这些激酶的信号转导决定了不同的细胞结果,包括细胞周期停滞、细胞死亡、细胞分化、应激适应和炎症。p38 和 JNK 在代谢调节中的强大作用,这已在广泛的条件性敲除 (KO) 小鼠模型中得到证实。这些研究表明,JNK 激酶与组织特异性和全身胰岛素敏感性的调节、肝脂质沉积和脂肪因子的产生等有关。在代谢调节中的众多作用中,p38 调节β细胞存活和脂肪组织中的关键过程,如产热和脂肪分解。p38 和 JNK 的激活与肥胖和代谢综合征有关,并且靶向这些激酶或其信号转导通路的非必需组分已被提议作为治疗或预防这些代谢性疾病的治疗方法。特别是,JNK 缺失小鼠对高脂肪饮食诱导的胰岛素抵抗和肝脂肪变性的保护 表明 MAP 激酶信号通路是代谢灵活性的关键调节因子和代谢下降的驱动因素。
MAP 激酶通过涉及上游 MAP 激酶激酶 (MAP2K) 和 MAP 激酶激酶激酶 (MAP3K) 的信号转导级联反应被激活。这些成分中最上游的MAP3K是一组21种人类激酶,我们只知道其中少数的激活机制和信号。最近引起人们兴趣的一种 MAP3K 是 ZAKα,它与核糖体相互作用,是翻译损伤的传感器。ZAKα 通过两个 C 末端核糖体结合结构域与核糖体结合,并通过核糖体停滞和/或碰撞等扰动激活。这种用于监测核糖体功能并将核糖体畸变转化为 p38 和 JNK 激活的通路称为核毒性应激反应 (RSR)。我们关于RSR功能的知识已经在哺乳动物细胞系中收集,其治疗包括核肉毒素酶(包括蓖麻毒素,志贺毒素和α-肌肽,所有这些都与人类毒素具有病理相关性),抗生素(包括茴香霉素和环己酰亚胺)或紫外线照射,分别破坏或化学抑制核糖体或损伤 mRNA 模板。除了皮肤角质形成细胞的 UVB 照射外,这些治疗既没有提供对核糖体停滞、核糖体碰撞和 RSR 激活的生理来源的见解,也没有提供任何关于该信号通路在整个生物体环境中的生理作用的线索。在这里,我们表明ROS是ZAKα和下游RSR信号传导的强大激活剂,并且产生ROS的致肥胖饮食与小鼠肝脏中核糖体伸长动力学的改变有关。在肥胖和衰老的背景下,RSR通路介导众所周知但不需要的代谢转化,如葡萄糖耐量失调和肝脂肪变性。我们的工作为MAP激酶信号传导的代谢调节提供了机制见解,并指出核糖体是迄今为止未被重视的代谢应激传感器。
结果
ROS 抑制蛋白质合成并激活 RSR
氧化应激和 ROS 均抑制翻译 并导致 p38 和 JNK 激活 。为了寻找这些效应之间的潜在联系,我们用p38激活剂量的甲萘醌处理U2OS细胞,甲萘醌通过徒劳的还原-氧化循环导致细胞内超氧自由基的产生(图1A)。p38 激活被 ZAK 激酶活性抑制剂和 CRISPR 介导的 Zak 基因失活完全消除(图 1B)。甲萘醌似乎激活了 RSR,因为p38 激活需要核糖体结合的 α 亚型,但不需要 ZAK 的无关β亚型(图 1、C 和 D)。为了进一步支持这一观点,我们发现甲萘醌在嘌呤霉素掺入测定中强烈抑制批量翻译(图 1E),并且 ZAKα 中需要功能性核糖体结合域才能激活 p38(图 1F)。这些影响是可逆的,因为当甲萘醌在ROS清除剂N-乙酰半胱氨酸(NAC)存在下被洗掉时,细胞既恢复了翻译又沉默了p38活性,尽管有一些延迟(图1G)。甲萘醌还激活了综合应激反应(ISR),如eIF2α磷酸化所证明的那样,上述所有效应都可以通过用NAC预孵育细胞来规避(图1E和图S1A)。ROS的这些作用不仅限于甲萘醌,因为我们发现氧化还原循环剂β-拉帕醌和硫氧还蛋白还原酶抑制剂金诺芬同样降低了核糖体输出,并以ZAK 依赖性和 NAC 可逆的方式激活了 p38 和 JNK(图 1,H 至 J,以及图 S1、B 至 D)。基于这些结果,我们得出结论,ROS暴露会急性触发细胞中的RSR信号传导。
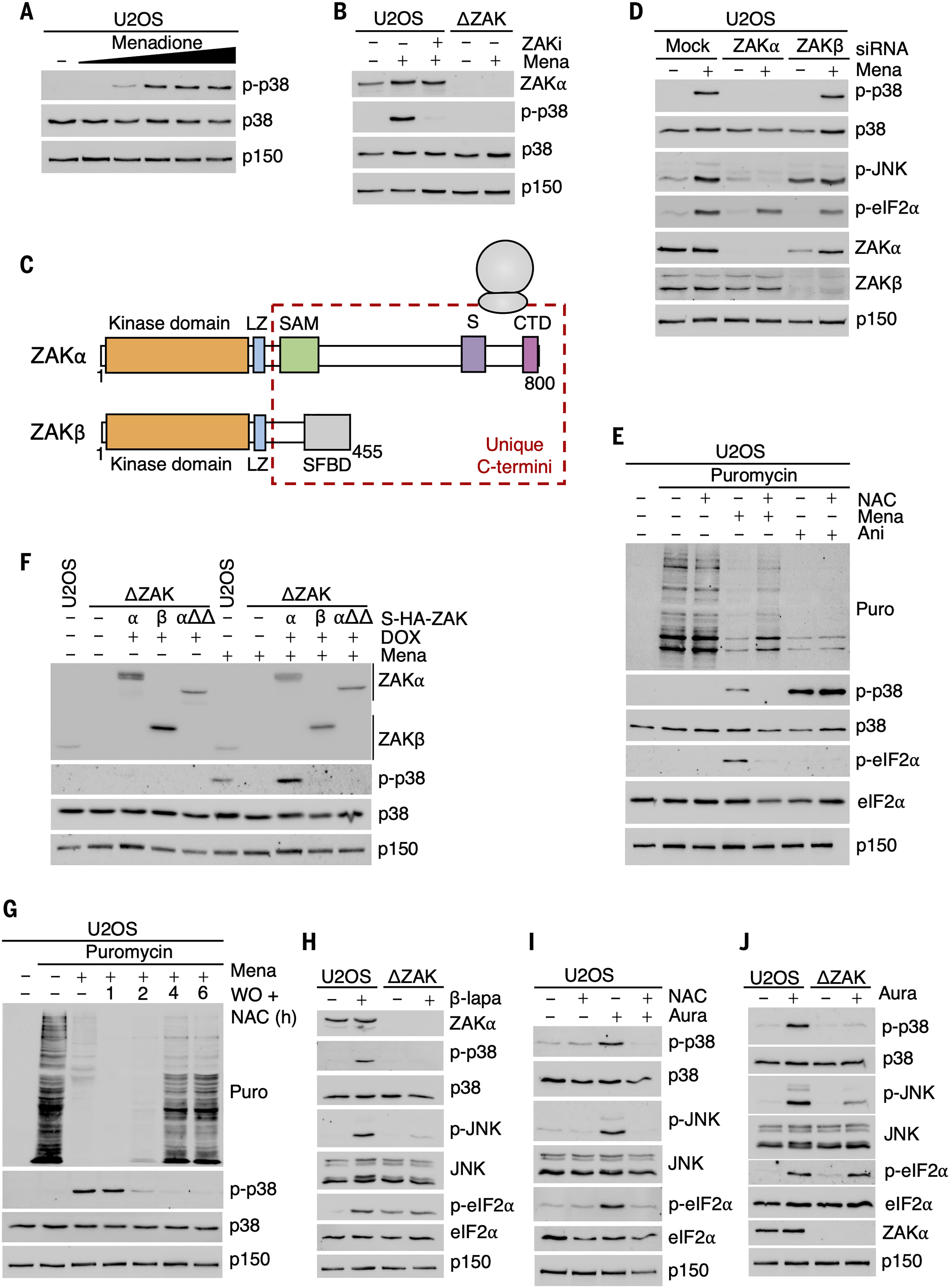
图 1.ROS 抑制翻译并激活 RSR。
(A) U2OS 细胞用浓度递增的甲萘醌(25、50、150、250 和 500 μM,持续 1 小时)处理。用指定的抗体通过免疫印迹分析裂解物。(B) WT U2OS 细胞或因 ZAK (ΔZAK) 而缺失的 U2OS 细胞用 ZAK 抑制剂 (ZAKi,2 μM) 和甲萘醌 (Mena, 250 μM) 处理 1 小时。裂解物分析如(A)所示。(C) ZAK蛋白亚型示意图。LZ, 亮氨酸拉链;SAM, 无菌α基序;S, 传感器域;CTD, C端结构域;SFBD,应力纤维结合域。(D) U2OS 细胞转染对照(模拟)小干扰 RNA (siRNA) 或靶向 ZAK α或β亚型的 siRNA。用甲萘醌(250μM,1小时)处理细胞,并分析裂解物,如(A)所示。(E) U2OS 细胞用 NAC(10 mM,1 小时)预处理,然后加入甲萘醌(250 μM,1 小时)或茴香霉素(Ani,1 μg/ml,1 小时)。在收获前10分钟将嘌呤霉素(10μg/ ml)加入培养物中,并通过免疫印迹用指定的抗体分析裂解物。(F) U2OS 细胞、ΔZAK 细胞和用 WT 和突变形式的 ZAKα 和 WT ZAKβ 救活的 ΔZAK 细胞按 (D) [αΔΔ 是 (C) 中“S”和“CTD”结构域的缺失处理]。裂解物分析如(A)所示。(G) U2OS 细胞用甲萘醌(250 μM,持续 1 小时)处理,然后在 NAC (10 mM) 存在下洗脱 (WO) 指定时间。在收获前用嘌呤霉素(10μg/ ml,10分钟)脉冲处理细胞,如(E)所示。裂解物分析如(A)所示。(H)用产生ROS的β-拉帕醌(β-lapa,20μM,1小时)处理(B)的细胞,并分析裂解物,如(A)所示。(I) U2OS 细胞用 NAC(10 mM,1 小时)预处理,然后按照指示加入金诺芬(Aura,5 μM,1 小时)。裂解物分析如(A)所示。(J)用金诺芬(5μM,1小时)处理(B)的细胞,并分析裂解物,如(A)所示。
ROS在体内诱导核糖体停滞,并在体外损害翻译
ZAKα 对核糖体的停滞和碰撞均有反应。为了了解 ROS 诱导的 RSR 激活的机制基础,我们通过用微球菌核酸酶 (MNase) 消化多核糖体并在蔗糖梯度上解析碰撞的核糖体来分析细胞中的核糖体碰撞。在用甲萘醌处理细胞 30 分钟或 1 小时后,这种方法没有显示 MNase 抗性多核糖体峰的增加,这表明核糖体堆积(图 2A 和图 S1E,左)。然而,用β-拉帕醌处理U2OS细胞30分钟后,碰撞的核糖体略有增加(图S1F,左)。这些结果与茴香霉素处理形成鲜明对比,茴香霉素处理导致大量核糖体碰撞(图 S1G),如前所述 。我们和其他人之前已经报道过,氨基酸剥夺会导致延长的核糖体停滞,但这些停滞的核糖体仅在抑制 ISR 时转化为碰撞。由于 ROS 也激活了 ISR(图 1),我们想知道这种反应是否也限制了对甲萘醌和 β-拉帕醌的核糖体碰撞。在加入甲萘醌和β-拉帕醌后30分钟,用泛ISR抑制剂ISRIB处理细胞确实刺激了低水平的核糖体碰撞(图2A和图S1F,右和H),但这些影响是短暂的,在1小时和2小时的时间点基本上无法检测到(图S1E)。这些数据表明,细胞对ROS生成剂的剧烈反应,核糖体停滞和碰撞,并且高水平的ROS本身会干扰以后时间点的翻译启动。事实上,甲萘醌和β-拉帕醌治疗都伴随着多核糖体的时间依赖性丢失,而ISRIB无法逆转(图S1,I和J)。如前所述,用沙丁氨酸靶向起始 80个 S 核糖体导致拉长核糖体的快速径流(图 2B,左),如前所示。甲萘醌处理的细胞中剩余的翻译核糖体的径流受到阻碍(图2B,右),进一步支持ROS减慢和/或停滞延长核糖体。与上述结果一致,与茴香霉素处理相比,来自甲萘醌处理的细胞的核糖体富集沉淀对碰撞伸长 [EDF1 、泛素化 RPS10 和 ZNF598 ] 和起始 [泛素化 RPS2 ] 核糖体(图 S2、A 和 B)。此外,在用甲萘醌和金诺芬处理细胞时,内质网 (ER) 应激反应激酶 PERK 而不是核糖体相关的 GCN2 似乎是更相关的 eIF2α 激酶(图 2C 和图 S2C)。这些和先前的数据表明,急性 ROS 暴露时 ZAKα 的激活可以独立于广泛的核糖体碰撞而发生,因此可能在很大程度上取决于单个核糖体的停滞和减慢(图 S2D)。我们的研究结果还强调,内质网应激和核毒性应激的触发因素是重叠的。事实上,内质网应激诱导剂thapsigargin强烈抑制了大量核糖体翻译并诱导了ZAKα的激活(图S2,E至G)。
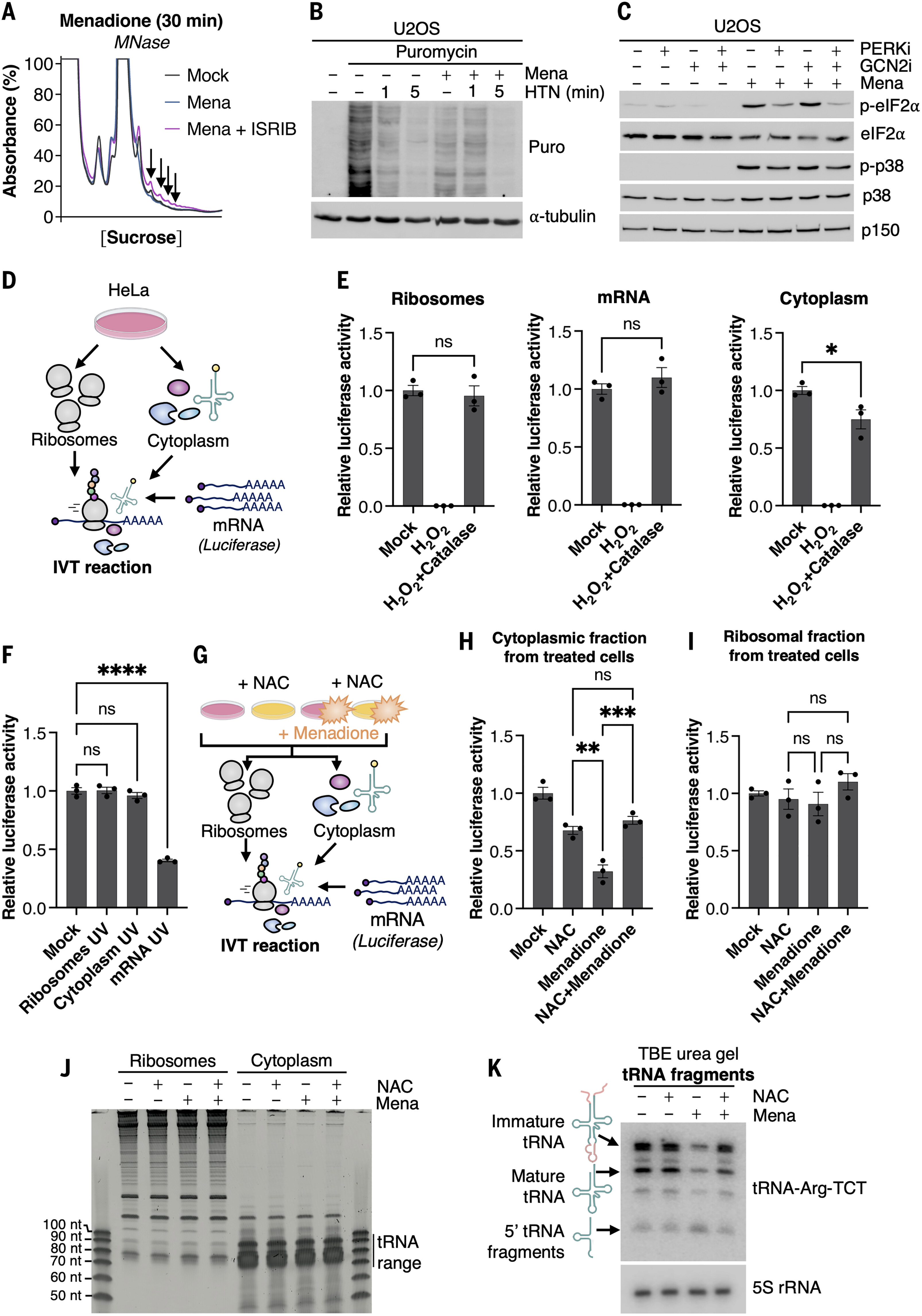
图 2.ROS 诱导的 ZAKα 激活与核糖体停滞和碰撞有关。
(A) U2OS 细胞用甲萘醌(Mena,250 μM,持续 30 分钟)和 ISRIB(200 nM)处理。裂解物用MNase消化,并在线性蔗糖梯度上分离。箭头突出显示表明核糖体碰撞增加的UV峰。(B) U2OS 细胞按指示用甲萘醌(250 μM,1 小时)和 harringtonine(HTN,2 μg/ml)预处理指定时间以诱导核糖体径流。在收获前(但在HTN孵育时间之后)将嘌呤霉素(10μg/ ml)加入培养物中5分钟,并通过免疫印迹用指示的抗体分析裂解物。(C) U2OS 细胞如图所示,用甲萘醌(250 μM,持续 1 小时)和抑制剂(i,1 μM)处理 PERK 和 GCN2。裂解物分析如(B)所示。(D) 三方体外翻译(IVT)方法示意图。核糖体、无核糖体细胞质和 mRNA 可以在联合使用前单独处理。(E)将(D)的三个馏分分别用过氧化氢(10分钟)处理,并如图所示通过添加过氧化氢酶中和。通过荧光素酶测定法测定联合反应中的翻译效率。(F)与(E)相同,除了馏分单独用UVB(500 J / m)照射2)在体外翻译之前。(G)改良的三方IVT测定示意图。与(D)相比,用NAC(10mM,1小时)预处理HeLa细胞,然后在纯化馏分之前加入甲萘醌(250μM,1小时)。(H) 用来自 (G) 的无核糖体细胞质处理的联合 IVT 反应。(I)与(H)相同,只是使用了来自(G)的处理过的核糖体。所有值均表示荧光素酶活性归一化为对照。对于 (E)、(F)、(H) 和 (I),数据绘制为平均值,所有误差线表示 SEM(n = 3 个生物学重复)。ns, 无显著性;*P ≤ 0.05;**P ≤ 0.01;市≤ 0.001;P ≤ 0.0001 通过两组学生 t 检验和两组以上 Tukey 事后检验的单因素方差分析。(J) 从(G)的HeLa细胞组分中分离出的全细胞RNA,在尿素琼脂糖凝胶上分离并进行RNA染色。NT,核苷酸。(K) (J) (J) RNA 样品上 tRNA-Arg-TCT 的 Northern 印迹。对应于不同条带的tRNA中间体的示意图显示在印迹的左侧。
ROS 有可能破坏 DNA 和 RNA 中的核苷酸,并且核糖体 RNA (rRNA) 和 mRNA 碱基的氧化修饰以前与翻译损伤有关。为了了解翻译过程中的哪些组分对ROS敏感,我们从HeLa细胞的裂解物中开发了一种三方体外翻译系统(图2D)。该细胞系还显示出甲萘醌诱导和NAC可逆的翻译抑制和p38激活(图S2,H和I)。我们的方法包括从一种HeLa细胞培养物中分离核糖体,以及从另一种培养物中分离含有转移RNA(tRNA)和起始因子和延伸因子的无核糖体细胞质部分,只有在组合时才支持体外转录的荧光素酶编码mRNA的翻译(图2D和图S2J)。为了将氧化损伤引入体外翻译系统,我们一次只用过氧化氢处理三种组分中的一种 10 分钟。为了防止残留效应,我们随后用过氧化氢酶中和过氧化氢,然后合并馏分并确定荧光素酶蛋白的产生程度。令我们惊讶的是,过氧化氢处理并未损害核糖体或荧光素酶mRNA支持翻译的能力,相反,细胞质部分对氧化损伤非常敏感(图2E)。如前所述,这一结果与引起碰撞的 UVB 照射形成鲜明对比,后者仅对 mRNA 成分产生负面影响 (图 2F)。我们还直接从甲萘醌和NAC处理的HeLa细胞中制备核糖体和细胞质组分(图2G)。在这些实验中,我们的结果表明,细胞质部分(而不是核糖体部分)含有一种或多种可溶性ROS敏感成分(图2,H和I)。使用生物素转换测定,我们没有发现甲萘醌处理后许多相关翻译起始和延伸因子中半胱氨酸氧化的生化证据(图 S3A)。对tRNA完整性的全局检查也没有表明细胞质部分中的tRNA库发生大规模降解(图2J)。然而,tRNA Arg-TCT 的 Northern 印迹显示明显的切割和碎裂,这是由甲萘醌诱导的,并被 NAC 逆转(图 2K)。对 tRNA Leu-CAA、Gly-GCC 和 iMet-CAT 观察到类似但弱得多的影响(图 S3,B 至 D),对 tRNA 电荷没有明显影响(图 S3E)。血管生成素是一种 tRNA 裂解 RNase,在大量细胞应激损伤时被激活,产生抑制翻译的 tRNA 片段 。最近证实了甲萘醌激活血管生成素并随后进行 tRNA 切割。尽管这些影响可能不是唯一相关的影响,但我们得出结论,ROS诱导剂会干扰体内和体外的翻译并激活ZAKα和RSR。
来自 ZAKα 和 ASK1 的贡献是 ROS 诱导的 p38 和 JNK 激活的基础
当用甲萘醌攻击时,p38 激活发生在两个不同的波中,一个在暴露后 5 分钟开始,在 10 至 15 分钟达到峰值,另一个在 45 分钟开始,在 1 至 2 小时达到峰值(图 3A)。这些峰中只有第二个峰与ISR激活、翻译关闭和氧化蛋白的出现相吻合(图3A和图S3F)。当单独或组合分析 ZAK 和 ASK1 基因缺失的 U2OS 细胞时,ROS 激活的 MAP3K ASK1 似乎仅参与这些反应中的第一个反应,而 ZAKα 仅参与第二波反应(图 3B)。这些数据表明,ASK1 对氧化应激的反应更快,并且对大分子的损伤和核糖体损伤先于 RSR 的激活。我们还用过氧化氢挑战我们的 U2OS 细胞,并观察到 ZAK 和 ASK1 激酶活性对应激相关 MAPK 激活的部分依赖性(图 S3G)。我们的研究结果增加了关于ASK1作为ROS激活的MAP3K的现有知识,并强调ZAKα是平行通路中的一个纽带,该通路将ROS诱导的翻译损伤传递给MAPK信号转导(图3C)。
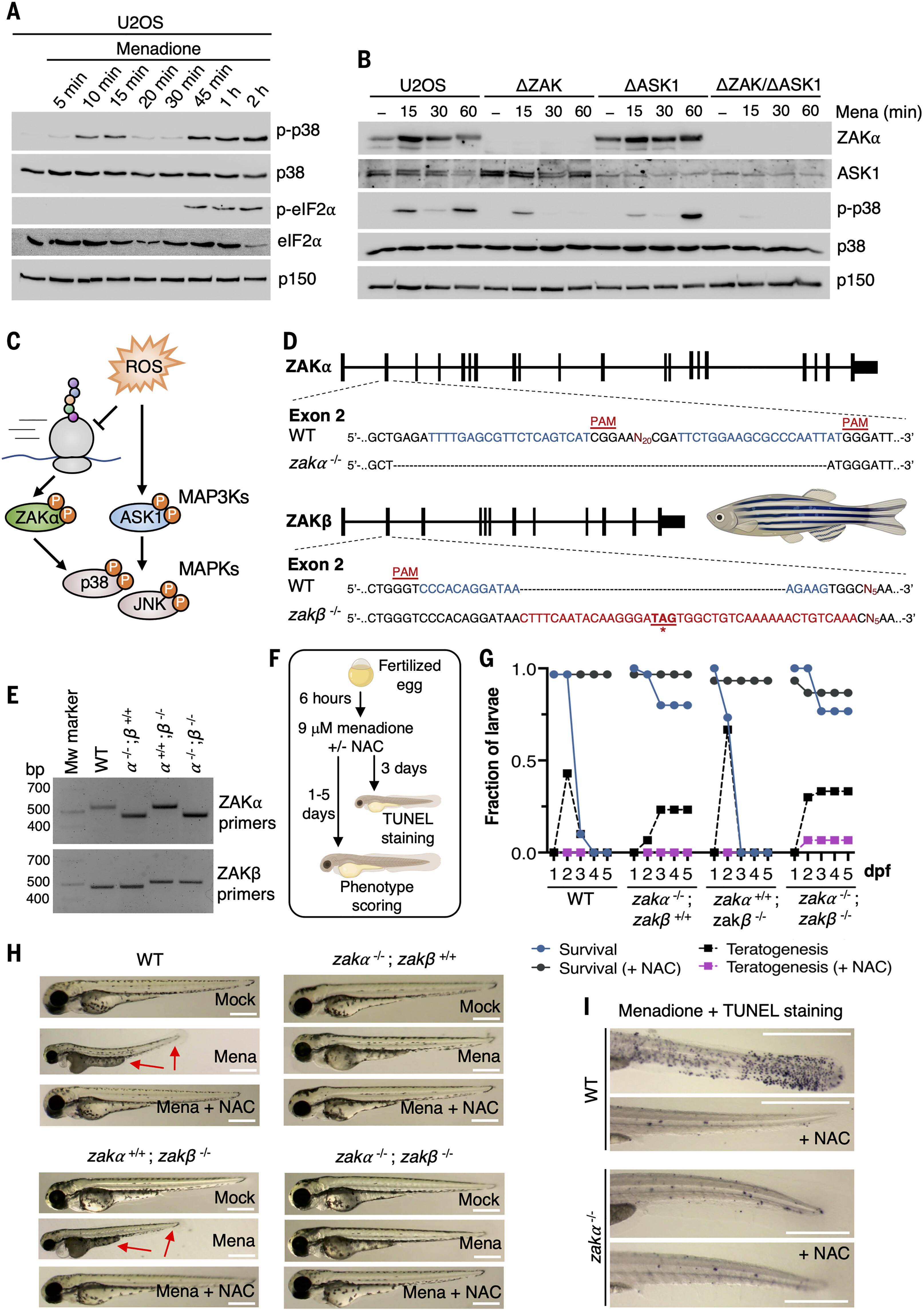
图 3.ZAKα介导甲萘醌诱导的斑马鱼凋亡和死亡。
(A) U2OS 细胞用甲萘醌 (250 μM) 处理指定时间。用指定的抗体通过免疫印迹分析裂解物。(B) 分别删除 ZAK (ΔZAK)、ASK1 (ΔASK1) 和两者 (ΔZAK/ΔASK1) 的 U2OS 细胞,用甲萘醌 (250 μM) 处理指定时间。裂解物分析如(A)所示。(C) ROS通过平行传感机制诱导p38和JNK激酶激活的模型。ZAKα 对 ROS 诱导的核糖体翻译损伤有反应(左图),而 ASK1 活性直接受细胞内 ROS 水平(右图)控制。(D) 斑马鱼 zakα(上)和 zakβ(下)基因外显子 2 中向导 RNA 序列(蓝色)和衍生敲除等位基因的基因组定位。突变 zakβ 中 33 bp 的插入以红色突出显示,框内 STOP 密码子的位置带有下划线和粗体。PAM,原间隔片相邻基序。(E) 来自(D)的zakα和zakβ基因的WT和CRISPR修饰等位基因的基因分型。裂解每个基因型的单个斑马鱼幼虫,并使用指定的引物对进行基因组PCR。通过琼脂糖凝胶电泳分离扩增的条带。(F)甲萘醌处理斑马鱼幼虫的实验示意图。斑马鱼卵在甲萘醌(9μM)和/或NAC(60μM)存在下从受精后6小时至5天孵育。每天对幼虫进行一次死亡或活体评分,并确定具有发育表型的鱼类数量。为了检测细胞凋亡,将处理3天的幼虫固定并进行TUNEL染色。(G)对具有每种指定基因型的30只斑马鱼幼虫进行处理和评分,如(F)所示。只有心脏骤停的幼虫被判定为死亡。显示的数据来自所进行的三个实验中的一个代表性实验。DPF,受精后天数。(H) 第 3 天 (G) 幼虫的代表性图像。用甲萘醌(Mena)处理的WT鱼表现为卵黄囊变黑、心脏水肿和短扭结的尾巴(红色箭头)。NAC的加入或zakα的缺失在很大程度上消除了这些病理。(I) WT和zakα−/−将幼虫按(F)处理3天,固定,并进行TUNEL染色。照片显示了幼虫尾部的代表性图像。所有比例尺,500 μm。
ZAKα介导ROS诱导的斑马鱼幼虫死亡
为了探究ROS诱导的RSR信号传导的有机体后果,我们转向斑马鱼作为模式生物。斑马鱼适合 CRISPR 介导的基因操作,并且由于其快速的宫外发育而成为发育研究的有吸引力的生物体。哺乳动物含有编码两种不同剪接变体的单个 Zak 基因(图 1C),而斑马鱼则表达来自两个独立基因的 ZAKα 和 ZAKβ 的清晰直系同源物(图 S3H)。因此,我们继续在斑马鱼中中断这两个基因(图.3D)并产生单KO和双KO动物(图3E)。遗传引入的缺失中断了两个旁系同源物的开放阅读框,也降低了它们的mRNA丰度,zakβ敲除还影响了zakα的表达(减少50%)(图S3I)。
受精野生型 (WT) 斑马鱼卵暴露于甲萘醌导致大多数幼虫在 3 至 4 天后死亡(评分为心脏骤停)(图 3、F 和 G)。在此之前,出现了一系列发育表型,例如卵黄囊和心脏的水肿、脊柱或尾巴弯曲以及长度缩短(图 3H)。所有这些显着的影响都可以归因于病理性ROS的产生,因为它们被NAC在培养水中的共施所阻止(图3,G和H)。虽然zakβ−/−鱼类对甲萘醌(Zakα−/−)的作用同样敏感双KO幼虫对死亡和病理表型的出现有很大抵抗力(图3,G和H)。甲萘醌诱导的病理学与凋亡细胞死亡有关,如WT幼虫尾部的TUNEL阳性细胞所观察到的那样,这种作用也可以通过NAC或zakα缺失的共同给药来预防(图3I)。我们得出结论,ZAKα,而不是 ZAKβ,在生物体环境中对 ROS 有反应,并且 zakα−/−至少在短期内,斑马鱼幼虫受到很好的保护,免受ROS病理性爆发的有害影响。
当喂食富含卡路里的饮食时,ZAKα−/−可以保护小鼠免受代谢功能障碍的影响
通过检查人体组织中 ZAKα 和 ASK1 转录本的相对表达水平,我们观察到肝脏含有相对较少的 ASK1 mRNA,同时也是 ZAKα 表达超过 ZAKβ 的极少数组织之一(图 S3J;数据来自 GTExPortal:https://gtexportal.org/)。 对小鼠蛋白质组最近组织分辨草稿的重新分析还表明,ZAKα 蛋白在包括肝脏在内的多个代谢器官中的表达水平高于 ASK1(图 S3K)。我们继续进行男性 WT 和 ZAK−/−小鼠在富含脂质和糖(高脂肪,高糖:HFHS)的饮食中持续25周,仅通过腹膜内葡萄糖耐量试验(ipGTT)和磁共振(MR)扫描中断以确定身体成分(图4A)。已知这种饮食会导致肥胖、胰岛素抵抗和肝脂肪变性增加,ROS 生成增加至少部分是潜在的病理驱动因素。正如我们之前报道的,雄性 ZAK−/− 的起始重量小鼠略低于WT同窝小鼠(图S4A)。ZAK−/−的初始体重增加也有所延迟小鼠从食物饮食转向HFHS饮食,但小鼠随后以基本与基因型无关的方式继续增加体重(图4B),并在整个实验过程中摄入相似量的食物(图S4B)。MR扫描显示,WT小鼠的脂肪量如预期的那样增加(图S4C),但在ZAK−/−中有一些延迟小鼠(图S4C),在两种情况下都具有相对恒定的瘦质量(图S4D)。在 WT 小鼠中,HFHS 喂养也导致 ipGTT 确定的预期血糖控制丧失,在 8 周时很明显,在 19 周时加剧(图 4C)。相反,HFHS 喂养的 ZAK−/−小鼠在8周的时间点对这种功能代谢下降有很好的保护,并且在ipGTT中的表现与食物喂养的小鼠相似(图4C,顶部与中部)。在 19 周的时间点,ZAK−/−小鼠似乎已经发展出一定程度的葡萄糖不耐受,尽管这不如WT明显(图4C,底部)。与此一致,ZAK−/−小鼠在 19 周的时间点表现出较少的胰岛素抵抗,HOMA-IR(胰岛素抵抗稳态模型评估)值降低 50% 证明(图 4D 和图 S4、E 和 F)。与葡萄糖耐受不良相反,通过HOMA-IR或在较长时间跨度内开发的胰岛素激发试验评估的明显胰岛素抵抗。与此一致,在不同的小鼠队列中,WT小鼠似乎对胰岛素敏感,因此与ZAK−/−HFHS 喂养 8 周后的小鼠没有显着差异(图 4E),因此不能完全概括 JNK 缺陷小鼠的报告表型。安乐死后测量的肝脏重量在 WT 和 ZAK −/−小鼠之间相似(图S4G),但在HFHS喂养的ZAK−/−中,肝脏甘油三酯含量降低~50%尽管小鼠血清甘油三酯水平难以区分(图4F和图S4H)。这种差异也表现为ZAK−/−的脂肪变性等级较低肝脏(图4,G和H)。
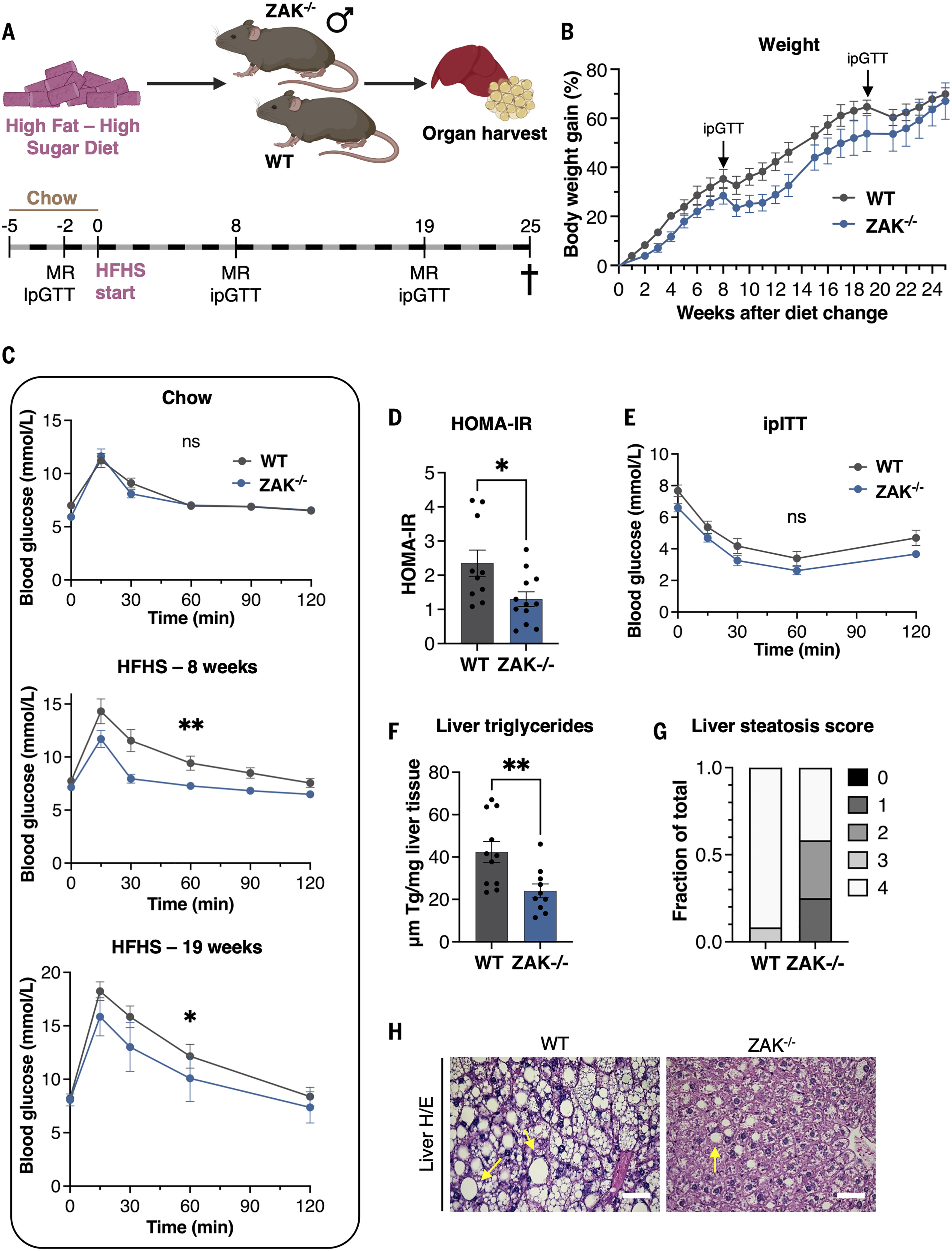
图 4.ZAKα−/−小鼠受到保护,免受肥胖相关的代谢功能障碍。
(A)小鼠喂养实验示意图。10 至 12 周龄男性 WT (n = 13) 和 ZAK−/− (n = 12) 将小鼠维持在食物上,并在转向 HFHS 饮食之前进行 MR 扫描和 ipGTT 2 周。小鼠在进一步 8 周和 19 周的 HFHS 喂养后进行 MR 扫描和 ipGTT。对小鼠实施安乐死,并在饮食转变后25周收集组织。(B)从(A)开始的小鼠体重增加百分比。箭头标记 IpGTT 测定的时间和由此产生的瞬时失重。(C)来自(A)的小鼠的血糖浓度,经ipGTT测定。(D)来自(A)的小鼠的HOMA-IR。(E)图中小鼠的血糖浓度。S4I 在 8 周时间点接受 IpITT (WT, n = 12;ZAKα−/−,n = 7)。(F)(A)小鼠肝脏甘油三酯含量。(G) 从 (A) 对小鼠的肝脂肪变性等级(量表,0 至 4)进行评分。(H)代表性苏木精和伊红(H&E)染色的WT和ZA−/−K的图像(A)中小鼠的肝脏切片。箭头表示脂肪变性区域。对于(B)至(F),所有数据均绘制为平均值,所有误差线均表示SEM。对于(C)和(E),统计分析基于每个实验组的曲线下面积(AUC)。
观察到对饮食诱导的葡萄糖耐量不良的早期保护和对 ZAK −/−肝脂肪变性的长期保护尽管在喂养 8 周时具有相似的全身胰岛素敏感性,但 WT 小鼠促使我们寻找 Zak 缺失提供的代谢保护的替代解释。首先,我们进行了 WT 和 ZAK−/−HFHS饮食小鼠10周,使用氚化2-脱氧-D-葡萄糖(2DG)进行葡萄糖示踪实验。小鼠在注射后1小时被安乐死,并在几个中枢和外周组织中测定放射性葡萄糖的摄取(图S4I)。与我们的葡萄糖激发实验(图4C)一致,放射性从ZAK−/−小鼠的血液中清除得更快(图S4J,左上角),尽管基因型之间对2DG的摄取及其在终点转化为2DG-6-磷酸(2DG6P)是相似的(图S4J,其余面板)。这些结果表明 WT 和 ZAK−/− 中葡萄糖摄取的总容量相等60 分钟后的组织,在较早的时间点具有潜在的动力学效应。其次,我们将两种基因型的小鼠分别喂食食物或HFHS6周,在代谢室中1周,并监测它们的运动,O2的消耗和二氧化碳的产生(图 S5A)。运动活动和能量消耗在很大程度上与基因型无关(基因型内方差 = 0.0022 kcal/h2,见图S5,B至D;对于基因型间方差 = 0.00027 kcal/h2,见图S5C)。然而,我们观察到 ZAK−/− 的呼吸交换比率显着增加当小鼠被喂食常规食物时,它们在WT上(图S5,E至G)。这一结果表明,在ZAK−/−小鼠中,碳水化合物比脂质更可作为能量来源,可能是由于这些动物的肥胖减少。该读数仅在食物喂养的小鼠中可观察到,并且HFHS诱导的呼吸交换比率的明显抑制超过了两种基因型,从而阻碍了对肥胖小鼠的进一步询问。
为了评估 ROS 对与 ZAK KO 相关的代谢表型的贡献,我们将 HFHS 喂养与饮用水中每升 10 克 NAC 补充剂相结合。在 5 周和 10 周时进行 IpGTT 测定,并在治疗 12 周后对小鼠实施安乐死(图 S6A)。在这个实验中,ZAK−/−小鼠的体重增加最初也被延迟了,并通过高剂量的NAC进一步减少(图S6B)。IpGTT 测定证实了 WT 小鼠葡萄糖耐量不良和 HOMA-IR 升高的进行性发展,而 ZAK−/−在HFHS喂养5周和10周后,小鼠被完全保护免受这种代谢下降的影响(图S6,C至G)。补充NAC完全消除了基因型之间在ipGTT(图S6、C和D)、空腹胰岛素水平(图S6F)和HOMA-IR(图S6G)方面的这些差异。相反,补充NAC仅部分改善了WT小鼠的肝脂肪变性等级,并且似乎没有提供与ZAK KO相同的保护(图S6,H和I)。我们的结果表明,由 ROS 和其他核糖体损伤来源激活的 RSR 信号至少介导了代谢适应肥胖饮食的某些方面。
磷酸化蛋白质组学分析揭示了 ZAK−/− HFHS喂养的小鼠肝脏中 MAPK 信号转导的失调
我们使用磷酸化蛋白质组学研究了喂食 HFHS 5 周并在黑暗期安乐死的小鼠肝脏中 p38 和 JNK 激酶的信号转导,此时肝脏 ROS 水平最大(图 5A)。我们鉴定了 ∼5500 个独特的蛋白质和 ∼12,500 个独特的磷酸化位点(表 S1 和 S2)。主成分分析(PCA)显示,样品在蛋白质水平上的分离很少(图S7A),但在磷酸化位点水平上,根据基因型和饮食进行了明确的分离和聚类(图5B)。我们使用该数据集研究了所有记录的 MAPK、MAP2K 和 MAP3K 组分上注释为调节性的磷酸化变化(图 S7B)。该分析揭示了 p38 和 JNK 信号级联的几个组分上激活相关磷酸化位点的 HFHS 和 Zak 依赖性调节。对所有条件下相关磷酸肽的更精细分析显示,在 HFHS 喂养 ZAK−/− 时,JNK2 (MAPK9-Y185) 的活化相关磷酸化降低但不是WT小鼠(图5C,图S7C和表S2)。然而,对相同肝脏样本的蛋白质印迹分析并未表明 ZAK −/−小鼠中 HFHS 诱导的 JNK 磷酸化降低(图5D)。WT 小鼠中 HFHS 诱导的部分 RSR 信号转导可能发生在脂肪变性肝细胞中,我们设法通过免疫组织化学 (IHC) 检测弱 p-JNK 阳性(图 S7D)。我们还进行了小鼠实验,其中我们喂食了 WT 和 ZAK−/−小鼠咀嚼或HFHS16周。这些小鼠在安乐死前挨饿过夜,并通过蛋白质印迹分析它们的肝脏的 p-JNK(图 S7E)。这种方法也未能突出HFHS相关JNK活性的失调,这可能是由于我们的样本量小(图S7F)所解释的。

图 5.单体和二体足迹的核糖体分析揭示了 HFHS 喂养时翻译景观的谨慎变化。
(A)小鼠肝脏蛋白质组学和磷酸化蛋白质组学分析的实验设计和工作流程。将指定基因型的 10 至 12 周龄小鼠喂食 chow 或 HFHS 5 周 (n = 4 至 6)。(B) 来自(A)的小鼠肝脏磷酸化蛋白质组学谱的PCA。(C) MAPK9 (JNK2) 上激活相关磷酸化位点的 MS2 数据得出的强度条形图。条形的高度表示测量值的平均值,误差条表示 SE。 MS 数据中的缺失值绘制为零值。(D)通过免疫印迹分析(A)的肝脏裂解物,并对p-JNK除以总JNK信号进行定量分析(右)。(E)体内单体和二体分析实验示意图。12 周龄雄性 WT 小鼠喂食正常食物或 HFHS 饮食 8 周 (n = 3)。(F) 使用前 500 个表达基因对个体小鼠的单核(闭环)和双环(开放环)足迹数据的 PCA。(G) 图S8C(橙色)中单体足迹(深灰色)、二粒足迹(浅灰色)和差异密码子位点最突出基因对累积总信号的贡献。突出显示了累积数据集中 Alb、Apoe 和 Rbp4 的位置。(H) 在食物喂养(灰色)和 HFHS 喂养(粉红色)条件下,Apoe mRNA 上单体和二体足迹的位置特异性 A 位点信号。在单核子和双核子轨道下方,描绘了差异密码子位点的位置,高度对应于比值比值,并使用与图S8C相同的颜色编码。(i) 与(h)相同,但针对Rbp4。(J) 从 (H) 放大 Apoe 的左侧突出显示区域,特别是在 HFHS 和邻近的差异密码子位点中具有高二分信号,与该区域的碰撞增加一致。(K) 与(J)相同,但为右侧突出显示的区域。(L) 与 (J) 和 (K) 相同,但对于来自 (I) 中 Rbp4 转录本的高二分位点。
肝核糖体分析揭示了HFHS治疗后高表达转录本的全局翻译和不同停滞和碰撞位点的影响
使用核糖体分析(ribo-seq),我们接下来研究了HFHS饮食诱导的表型在多大程度上伴随着肝脏的翻译改变(图5E)。我们从两种足迹物种中制备了 ribo-seq 文库:标准的 ~30 nt 单体保护足迹和 ~60 nt 二核糖体足迹,它们表明核糖体堆积和碰撞 (54–56)。即使在生理条件下,也可以在小鼠肝脏和特定位置观察到丰富的二重足迹,这可能是暂时性翻译减慢位点的诊断,这些位点不会引起碰撞反应,例如 RSR。在我们的分析中,我们旨在确定加剧的双层覆盖率或依赖于HFHS的新位点。首先,来自前 500 个表达基因的足迹信号上的 PCA 表明按文库类型(单体或双体)分离,然后是饮食(chow 或 HFHS)(图 5F)。单体足迹分析表明,HFHS 饮食与对延伸动力学的强烈系统性、全转录组影响无关,如编码序列的 5' 和 3' 末端的足迹比对(图 S8A)和密码子停留时间分析(图 S8B)。接下来,我们将分析扩展到翻译组上的特定位点。我们使用最近报告的暂停评分指标 (58, 59) 计算了整个转录组范围内的位置特异性核糖体占有率,产生了 150 个位点增加和 160 个位点相对核糖体占有率降低 HFHS 与食物喂养的动物(图 S8C,左图)。其中许多位点对应于翻译组上具有高核糖体占有率的位置(图 S8C,右图),因此我们探索了影响翻译体积非常高的转录本上相对较少位点的翻译改变可能与 HFHS 治疗相关的可能性。因此,我们量化了单个高表达基因如何促进肝脏的整体翻译活性,这揭示了对一些显性转录本的强烈偏向,特别是编码血液分泌蛋白的mRNA,如白蛋白(Alb,占所有肝脏翻译的8.9%)、载脂蛋白E(Apoe,4.2%)和其他mRNA(图5G),深灰色曲线;119个基因占所有肝脏翻译的50%)。发现二分足迹具有全局相似的分布(图5G,浅灰色曲线)。差异密码子位点组对高度显性转录本的偏向性更强,其中 Alb、Apoe 和 Tfr 三种 mRNA 占检测到位点的一半(图 5G、橙色曲线和表 S3)。为了跟进这一观察结果,我们检查了排名靠前的转录本中的单核糖体和二核子覆盖率以及差异密码子位点。这些分析表明,HFHS和食物喂养动物之间的总体足迹分布高度相似且可重复,如Alb mRNA上的单核和双核覆盖模式所示(图S8D)。然而,对于一些高丰度的 mRNA,例如 Apoe(图 5H)和视黄醇结合蛋白 4(Rbp4;图 5I),我们在 HFHS 肝脏中特异性地鉴定了强烈富集的二重位点(图 5,H 和 I,橙色阴影和箭头)。对这些转录区域(图 5,J 到 L)的仔细检查表明,这些二分体位置位于上游,靠近单体足迹比值比分析中增加的差异位点,该星座与这些位点的碰撞增加相容。
ZAKα−/−小鼠在衰老过程中受到保护,免受代谢下降的影响
雄性小鼠在衰老过程中产生胰岛素抵抗,在非常老的年龄可以通过胰岛肥大来补偿。鉴于 ZAK −/−的显着保护在对抗肥胖代谢功能障碍的小鼠中,我们允许两性食物喂养的小鼠达到 14 至 16 个月大,然后再对它们进行 ipGTT(图 6A)。在这个年龄,与年轻小鼠相似,WT雄性比Zak缺失的对应物略重(图S8E),而雌性小鼠的体重与基因型无关(图S8F)。正如预期的那样,老年雄性WT小鼠在ipGTT测定中表现出强烈的血糖控制受损(图6B,顶部),而老年ZAK−/−的性能在该测定中,雄性小鼠与年轻雄性小鼠无法区分(图4C,顶部和图6B,顶部)。如前所述,雌性小鼠没有出现明显的衰老诱导的代谢下降(图6B,底部)。防止ZAK−/−雄性小鼠中与衰老相关的血糖控制下降的保护作用通过降低空腹血糖和胰岛素以及较低的HOMA-IR值进一步强调(图6,C至E)。与早期(8周)HFHS诱导的肥胖类似(图4,C至E),男性的这些变化发生在通过胰岛素耐量试验评估的全身胰岛素敏感性没有可测量的差异的情况下(图S8G)。对这些小鼠肝脏的检查显示,七只WT小鼠中有三只存在不同程度的肝脂肪变性,而六只ZAK−/−小鼠中只有一只小鼠存在不同程度的肝脂肪变性表现出最轻微的这种代谢疾病标志(图6,F和G),从而反映了图4中报道的饮食诱导肥胖的基因型特异性特征。

图 6.雄性ZAK−/−老化小鼠的健康代谢。
(A)小鼠衰老实验示意图。WT(雄性,n = 7;雌性,n = 16)和 ZAK−/−(雄性,n = 7;雌 性,n = 12)小鼠保持正常的食物饮食,直到14至16个月大(“老组”)。小鼠接受ipGTT并在2周后实施安乐死以收集组织。6 个月大的 WT 和 ZAK −/−雄性小鼠的组织(“年轻组”)用于比较。(B) 来自 (A) 的“老年”雄性(顶部)和雌性(底部)小鼠的血糖浓度,接受 ipGTT 测定。统计分析基于每个实验组的AUC。(C至E)来自(A)的禁食“老年”雄性小鼠的基础葡萄糖水平(C),基础胰岛素水平(D)和HOMA-IR(E)。(F)(A)中雄性小鼠的代表性H&E染色肝脏切片的图像。箭头表示脂肪变性区域。(G) 从 (A) 对雄性小鼠的肝脂肪变性等级(量表,0 至 4)进行评分。(H)(A)中雄性小鼠BAT的代表性H&E染色切片的图像。箭头表示明显的脂滴凝结和 BAT 美白的区域。(I)根据脂滴对(图S10A)的整个安装BAT扫描进行分割。适用于所有老化 WT 和 ZAK −/−的单独液滴尺寸计算样本并以直方图的形式表示 (n = 6)。(J)RSR对雄性小鼠肥胖和衰老的代谢调控模型。由于 HFHS 喂养或在衰老过程中产生的 ROS 会损害核糖体翻译并激活 RSR 传感器 ZAKα。下游 MAP 激酶(p38 和 JNK)信号转导介导代谢下降的特征,例如葡萄糖耐量不良、肝脂肪变性和 BAT 变白。
这些结果表明,肥胖和衰老中潜在的代谢应激信号存在重叠,尽管其幅度和持续时间不同。为了比较两种情况下肝脏氧化负荷的程度,我们进行了IHC以评估年轻与老年以及食物喂养与HFHS喂养的WT小鼠肝脏中4-HNE的含量。4-HNE是脂质过氧化的常用标志物,在诊断NASH和其他氧化应激相关疾病方面具有预后价值。尽管在食物喂养的年轻肝脏中对 4-HNE 进行 IHC 分析仅导致均匀的背景染色(图 S9A),但对图 4A 中 HFHS 喂养的小鼠的相同分析显示,整体染色显着增加,以及门静脉周围脂肪变性区域的典型富集(图 S9B)。在老年小鼠的肝脏中没有观察到这些模式(图S9C)。该分析表明,氧化应激负荷在老年小鼠肝脏中增加,但程度低于HFHS喂养的小鼠。
棕色脂肪组织结构和功能的恶化在ZAK−/−小鼠中减弱
雄性小鼠和人类的脂肪组织经历衰老诱导的退化,包括棕色脂肪组织 (BAT) 的“变白”。与老年WT小鼠的BAT切片相比,老年ZAK−/−小鼠的染色强度更接近于年轻小鼠的BAT(图S10A,以及图S11A)。这些差异在微观水平上也很明显,与来自ZAK的样本相比,多房BAT向更单房WAT样组织的转变在老年和年轻的WT−/−小鼠样本中更为明显(图6,H和I)。尽管影响通常较小,但在重新检查我们的 25 周 HFHS 队列时,我们观察到类似的 BAT 保护作用(图 4A;图 S10;和图 S11,A 和 B)。这些结构差异促使我们通过对剃光小鼠肩胛间BAT的红外成像来评估功能性BAT产热。尽管HFHS喂养与WT小鼠BAT温度的预期降低以及产热有关,但ZAK−/−小鼠与WT小鼠的产热有关在该组织中保持高温(图S11,C和D)。
讨论
在这里,我们表明ROS在体外,人类细胞系和小鼠中有效地干扰核糖体功能。这些畸变激活了 RSR 通路,最终导致氧化应激诱导的 p38 和 JNK 激酶激活。这种反应与病理生物体对氧化应激的反应(斑马鱼实验)和对增加的ROS产生的生理适应都有明显的相关性。在小鼠中,ROS诱导的RSR信号转导至少是一些对高脂肪饮食诱导的肥胖和衰老的代谢适应的基础,例如血糖控制的丧失和肝脂肪变性的发展。ZAK−/−保护喂食HFHS饮食时小鼠抵抗代谢功能障碍强调了这一点,以及在老年衰老ZAK−/−雄性小鼠中观察到的健康代谢(图6J)。上述见解强调核糖体损伤是一种重要的代谢应激信号,用于在生物体水平上调节新陈代谢。核糖体非常适合在广泛的背景下发挥温和应激信号支架的作用。由于它们在每个细胞中都大量存在,并且不断发挥其功能,因此仅少数核糖体的持续合成能力受损即可提供高度灵敏和快速的细胞扰动读数。先前的工作强调 mRNA 的氧化损伤是翻译损伤的来源,但我们的体外翻译实验强调可溶性细胞质因子是更敏感的组分。这些可能包括 tRNA 以及起始因子和延伸因子。虽然我们没有获得后者氧化损伤的证据,但我们证实了最近发表的 ROS 诱导的 tRNA 切割和片段化的证据。除了代表关键翻译组分的耗竭外,这些片段(可能由血管生成素RNase产生)通常被认为对核糖体功能具有抑制作用 。
JNK 信号转导可促进肥胖小鼠的胰岛素抵抗和肝脂肪变性。具体而言,当喂食高脂肪饮食时,肝脏中有条件地缺失 JNK1 和 JNK2 的小鼠可以免受这些结果的影响。相关的 JNK 靶标之一是 RXRα,其磷酸化可抑制 PPARα 复合体和 Fgf21 基因的转录激活。FGF21 是一种代谢应激诱导的激素,对胰岛素敏感性和肝脏肥胖均具有有益作用,这些作用在 jnk 缺失小鼠中会加剧。相反,髓系室中 p38 激活缺陷的小鼠在肝脏 FGF21 生成方面存在缺陷,并且在喂食高脂肪饮食时对代谢功能障碍敏感。一幅图景正在出现,其中 p38 和 JNK 信号可能根据组织、细胞类型和代谢环境竞争相反的结果。这些复杂性可能允许一种微调的代谢调节模式,这种模式共同体现在一个缓冲性极好的系统中。出乎意料的是,ZAKα−/−小鼠在中间时间点(8至10周)明显没有增加胰岛素敏感性的情况下,受到保护,免受代谢功能障碍的关键方面的影响。其他解释可能包括刺激胰岛素非依赖性葡萄糖摄取机制和/或改变能量来源的利用。
MAP3K ASK1 是细胞系和小鼠模型中 ROS 诱导的 p38 和 JNK 的主要激活剂 ,并且还与代谢调节有关。其他与代谢控制有关的 ROS 激活的 MAP3K 是 MLK2/3 和 MAP3K4。在这项工作中,我们描述了 ZAK KO 小鼠对 HFHS 诱导的葡萄糖耐量不良和肝脂肪变性的明确保护,并且至少其中一些表型与 ROS 介导的 ZAKα 和 RSR 激活有关。在 ZAKα 和 ASK1 在 ROS 诱导的 p38 和 JNK 激活中相互作用的一个合理模型中,ASK1 是 ROS 病理水平突然爆发时更相关的激酶,而 ZAKα 通过其感知翻译损伤的能力,对细胞内 ROS 轻微升高的影响更敏感。此外,这两种激酶(以及 MLK2/3、MAP3K4 和潜在的其他激酶)之间的分工可能是组织特异性的,因为 ZAKα 水平在器官之间尤其存在很大差异。应该注意的是,这里使用的实验系统在ROS暴露的来源、持续时间和幅度上有很大差异。总的来说,我们发现 ZAKα 作为 p38 和 JNK 激活和生物体保护的主要诱导剂的作用在我们的实验中最为明显,这些实验表明细胞系和斑马鱼的 ROS 水平过高。在长期暴露于较低水平的ROS(小鼠肥胖和衰老)的情况下,如上所述,替代机制可能发挥同样重要的作用。需要额外的工作来绘制 ROS 激活的 MAP3 激酶的完整清单及其与 ROS 驱动的生理和病理反应的相关性。
除了葡萄糖耐受不良和随机肝脂肪变性之外,ZAK−/−雄性小鼠免受BAT变白和肥胖和衰老时BAT完整性的破坏也至少部分受到保护。这些发现表明,RSR通路施加的代谢控制并不局限于肝脏,并且几种组织可能有助于ZAK−/−小鼠的整体代谢表型。事实上,最近的一份报告描述了 Zak 基因在促进亮氨酸限制小鼠小肠干细胞可塑性方面的独立作用。进一步增加了我们当前小鼠模型的复杂性,ZAKβ 亚型通过体积细胞压缩激活,并防止小鼠和人类的肌肉病变 ,并且这种剪接变体在全身 ZAK KO 动物中也被中断。在本研究中,没有实现Zakα亚型条件性KO小鼠模型的生成。未来朝这个方向所做的努力对于揭示RSR对我们描述的代谢表型的组织特异性贡献至关重要。我们也没有设法检测肝脏、脂肪组织和其他代谢相关器官中 p38 和 JNK 激酶亚型的 Zak 依赖性激活。我们怀疑潜在的技术解释是在HFHS喂养过程中这些应激激酶的长时间但微弱的过度激活,由于小鼠间差异大,传统方法很难检测到。尽管 p38 和 JNK 是 RSR 中公认的效应子,但体内缺乏从 ZAKα 到这些激酶的直接信号转导链接,这限制了我们对 ZAK KO 小鼠代谢保护机制的结论。最后,我们不知道我们在小鼠身上的发现在多大程度上可以外推到人类身上。在 ZAKα 和 ZAKβ 的共同激酶结构域中出现无义突变的患者表现为严重的早发性肌病,但没有代谢性表型的报道。然而,本研究中使用的小鼠喂养范式通常被认为是人类肥胖和相关代谢并发症的良好模型。在人类中,Zakα mRNA在所有主要代谢组织中都有稳健的鉴定,因此我们认为RSR也可能介导我们自己物种的代谢适应。
总之,我们的工作揭示了核糖体作为代谢调节信号转导平台的作用,并强调了RSR通路作为与肥胖和衰老相关的代谢变化的重要诱导剂。这些发现可能为开发对抗代谢性疾病的治疗策略提供机会。
该团队2022年发表的相关研究
Cell Metabolism | 核糖体停滞是通过核糖毒性应激反应进行代谢调节的信号
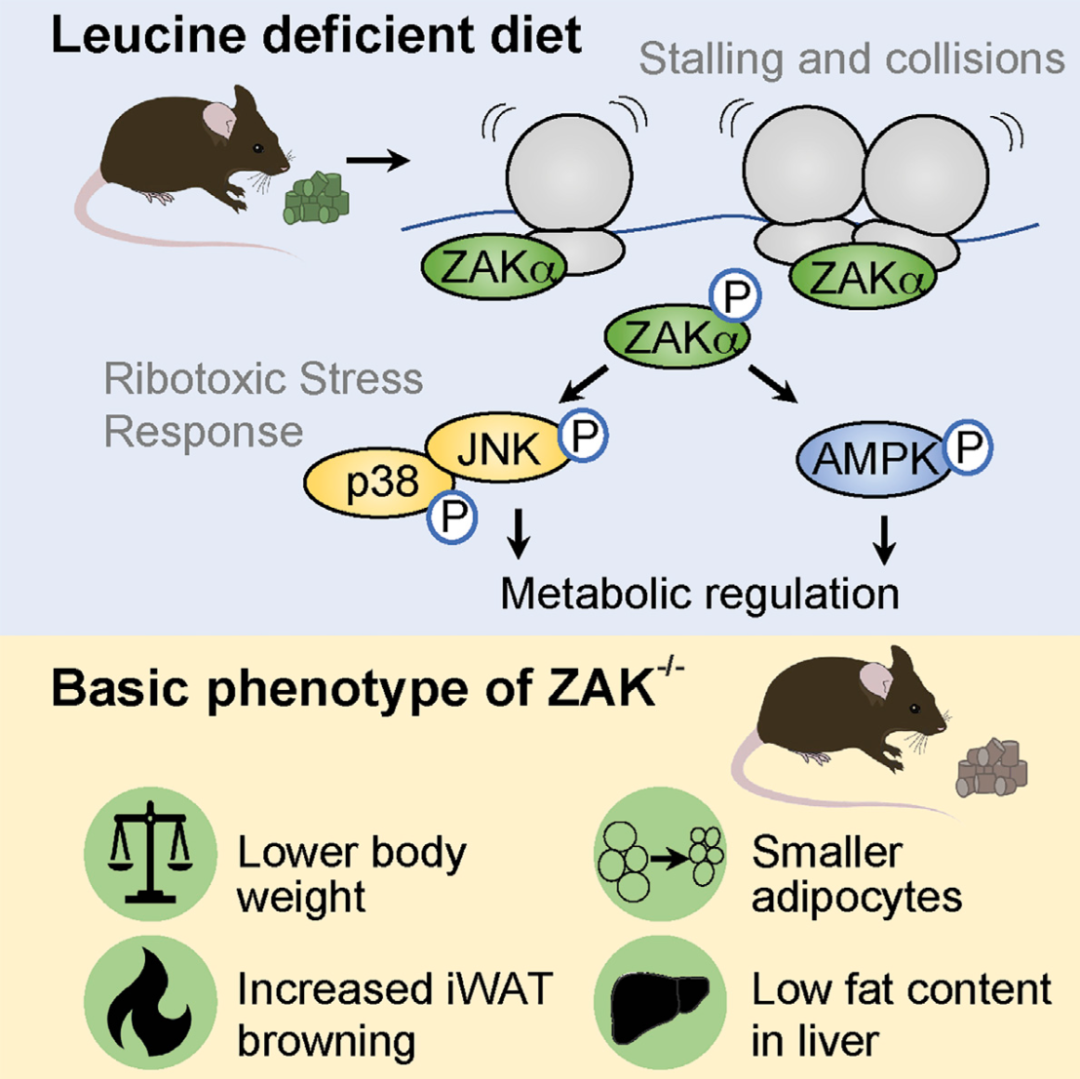
翻译的损伤可导致核糖体的碰撞,促进多个核糖体应力监测通路的激活。其中包括核毒性应激反应(RSR),MAP3K-ZAKα的核糖体感应导致p38和JNK激酶的激活。但关于核糖体损伤和下游RSR信号通路的生理影响仍然还不清楚。本研究建立氨基酸剥夺和营养饥饿模型,发现核糖体的停滞足以激活ZAKα,强调受损的核糖体作为代谢信号,并证明RSR信号在代谢调节中的作用。
1. 首先研究者对人类U2OS和HeLa细胞在厄尔平衡盐溶液(EBSS)进行饥饿培养,发现p38和JNK显著激活,而这些作用在相应的ZAK敲除细胞中被消除,证实RSR是由饥饿诱导的核糖体停滞激活的。并分析饥饿培养后mTOR底物S6激酶和4EBP1的磷酸化状态,发现ZAKα在代谢应激反应中调节AMPK和mTOR活性。
2. 对缺乏ZAK的模型生物进行饥饿反应研究,线虫与WT N2菌株相比,缺乏RSR组分zk-1和pmk-1在饥饿培养基中存活的时间更短。线虫中激酶之间的互作关系与在人类细胞中观察到的类似。
3. 对小鼠进行亮氨酸饥饿培养, 通过qPCR、ELISA、切片染色、激素和血糖调控等分析发现RSR信号在小鼠肝脏特异性和全身代谢适应氨基酸饥饿中都有作用,且RSR信号的遗传消融与ZAK−/−雄性小鼠的瘦表型相关,其特征是增加脂质周转和减少脂肪沉积。
4. 进一步证实在细胞饥饿和氨基酸缺乏的情况下,停滞的核糖体和在较小程度上碰撞的核糖体构成了激活ZAKa的潜在代谢信号。
参考文献
Ribosome stalling is a signal for metabolic regulation by the ribotoxic stress response. Cell Metabolism. 2022.
https://blog.sciencenet.cn/blog-41174-1412991.html
上一篇:肿瘤影响肾脏功能的激素效应
下一篇:猪羊等家畜动物们都拥有丰富的内心世界!
