ВЉЮФ
ЗЂЯжаТЕФЕААзжЪПеМфНсЙЙ
|
ЕААзжЪНсЙЙРрЫЦгюжцЕФАЕЮяжЪЃЌЮвУЧжЊЕРетжжЮяжЪДцдкаэЖрПЩФмЕФПеМфНсЙЙЃЌЕЋЮвУЧВЛжЊЕРОпЬхгаФФаЉНсЙЙЁЃРћгУAlphaFoldЩёОЭјТчЖдвкЭђИіЕААзжЪНсЙЙЛњаЕФмдЄВтЃЌШЛКѓДгетаЉдЄВтЕФНсЙЙжаЪЖБ№ГіаТаЭНсЙЙФЃЪНЃЌетПЩФмГЩЮЊЗЂЯжаТНсЙЙаТЙІФмЕФживЊПЊЪМЁЃдЄВтЮДРДдНРДдНЖраТЕФЕААзжЪЙІФмНсЙЙПЩФмЛсБЛЪЖБ№ГіРДЁЃ
баОПШЫдБвбОЭкОђСЫвЛИіАќКЌМИКѕУПИівбжЊЕААзжЪНсЙЙЕФЪ§ОнПтЃЌЪЙгУGoogle DeepMindЕФИяУќадAlphaFoldЩёОЭјТчдЄВтСЫГЌЙ§2вкИіНсЙЙЁЃетЯюЙЄзїНвЪОСЫЭъШЋаТЕФаЮзДЁЂЩњУќЛњаЕжаСюШЫОЊбШЕФСЊЯЕЃЌвдМАМИФъЧАЛЙЮоЗЈЯыЯѓЕФЦфЫћЖДВьЁЃ
FoldseekЮЊAlphaFoldЕААзжЪЪ§ОнПтЬсЙЉСЫвЛИіПьЫйЫбЫїЙЄОпЁЃ ЁАИааЛAlphaFoldЃЌЮвУЧЯждкПЩвдЬНЫїЮвУЧвдЧАвЛЮоЫљжЊЕФећИіЕААзжЪМвзхЃЌЁБЮїАрбРАЭШћТоФЩКЮШћХхЁЄПЈРзРДЋШОВЁбаОПЫљЃЈIJCЃЉЕФМЦЫуЩњЮябЇМвАЎЕТЛЊЖрЁЄВЈЫўХСЖћЖрЃЈEduard Porta PardoЃЉЫЕЃЌЫћУЛгаВЮгыНёФъ9дТ13ШедкЁЖздШЛЁЗЩЯЗЂБэЕФвЛЦЊТлЮФЁЃ
ШЅФъЃЌЙШИшDeepMindЪЙгУAlphaFoldдЄВтРДздОпгаЛљвђзщЪ§ОнЕФЩњЮяЬхЕФМИКѕЫљгавбжЊЕААзжЪЕФНсЙЙЃЌдкAlphaFoldЪ§ОнПтжаЛ§РлСЫдМ2.14вкжжНсЙЙЃЌИУЪ§ОнПтгЩгЂЙњаРЖйЕФХЗжоЗжзгЩњЮябЇЪЕбщЪвЕФХЗжоЩњЮяаХЯЂбЇбаОПЫљЃЈEMBL-EBIЃЉЭаЙмЁЃ
ЕААзжЪНсЙЙдЄВтЕФПЦбЇМвУЧЗЂЯжетИізЪдДЗЧГЃЗНБуЃЌЕЋЦфжааэЖрШЫжЛВщПДСЫвЛжжНсЙЙЃЌЛђепЪЧгыжЎЯрЙиЕФНсЙЙМвзхЃЌЪзЖћЙњСЂДѓбЇЕФМЦЫуЩњЮябЇМвТэЖЁЁЄЫЙЬЙФсИёЖћЃЈMartin SteineggerЃЉЫЕЃЌЫћЖдгГЩфећИіЪ§ОнПтЕФЙиЯЕИааЫШЄЁЃ ЁАЮвШЯЮЊПДПДЮвУЧЕФНсЙЙгюжцецЕФгаЖрДѓЪЧКмгаШЄЕФЁЃЁБ
ЮЊСЫзіЕНетвЛЕуЃЌЫЙЬЙФсИёЖћКЭМЦЫуЩњЮябЇМвХхЕТТоЁЄБДЬиРТоЃЈPedro BeltraoЃЉдкШ№ЪПЫеРшЪРЕФETH ZurichСьЕМЕФвЛИіЭХЖгПЊЗЂСЫвЛИіЙЄОпЃЌИУЙЄОпПЩвдПьЫйБШНЯЪ§ОнПтжаЕФУПжжНсЙЙЃЌЛљгкЫќУЧаЮзДЕФЯрЫЦадЁЃетдкAlphaFoldЪ§ОнПтжаЪЖБ№СЫГЌЙ§200ЭђИіаЮзДЯрЫЦЕФЕААзжЪЁЎДиЁЏЁЃ
ДЋЭГЩЯЃЌбаОПШЫдБЪЙгУЛљвђБрТыЕФЕААзжЪађСаНјааДЫРрБШНЯЁЃЕЋЪЧгыЫќУЧЕФНсЙЙЯрБШЃЌЕААзжЪађСадкНјЛЏЪБМфФкИќШнвзЗЂЩњБфЛЏЃЌЯожЦСЫевЕНЗЧГЃдЖдЕЯрЙиЕААзжЪЕФФмСІЁЃЫЙЬЙФсИёЖћЙРМЦЃЌЭЈЙ§БШНЯЕААзжЪНсЙЙЃЌЫћУЧЗЂЯжЕФгаЙиЕААзжЪДиЕФЪ§СПЪЧНіЪЙгУађСаЪБЕФЪЎБЖЁЃ
етаЉбаОПШЫдБИеИеПЊЪМЬНЫїетаЉдкаТЗЂЯжЕФЁЎаЧЯЕЁЏжаЃЌЕЋвбОГіЯжСЫвЛаЉСюШЫОЊбШЕФСЊЯЕЁЃР§ШчЃЌЫћУЧЗЂЯжвЛжжШЫРрКЭЦфЫћИДдггаЛњЬхгУРДМьВтВЁЖОDNAВЂДЅЗЂПьЫйУтвпЙЅЛїЕФЕААзжЪдквЛИігыЕЅЯИАћЯИОњКЭЙХОњЕФЕААзжЪДижаЁЊЁЊетЪЧвЛИіДЫЧАЮДжЊЕФСЊЯЕЃЌЫЙЬЙФсИёЖћЫЕЁЃ
ЖдгкГЌЙ§Ш§ЗжжЎвЛЕФЕААзжЪДиЃЌФПЧАМИКѕвЛЮоЫљжЊЁЃЁАЮвецЕФЯЃЭћЩњЮябЇМвФмНвЪОетИіКкАЕжЎДІЕФвЛаЉЙтУїЃЌЁБЫЙЬЙФсИёЖћЫЕЁЃ
ДгЮДМћЙ§ЕФаЮзД ЕкЖўИіЭХЖгВЩШЁСЫЩдЮЂВЛЭЌЕФЗНЗЈРДНвЪОЕААзжЪгюжцЕФАЕЮяжЪЁЃШ№ЪПАЭШћЖћДѓбЇКЭШ№ЪПЩњЮяаХЯЂбЇбаОПЫљЕФМЦЫуЩњЮябЇМвЧЧАВФШЁЄХхРзРЃЈJoana PereiraЃЉЁЂеВФЯЁЄЖХРбЧЖћМЊЃЈJanani DurairajЃЉЁЂЭаЫЙыјЁЄЪЉЮЄЕТЃЈTorsten SchwedeЃЉЕШШЫДДНЈСЫвЛИіСЌНгСЫ5000ЖрЭђИідкAlphaFoldЪ§ОнПтжазюОЋШЗдЄВтЕФНсЙЙЕФЭјТчЃЈИУЙЄОпЬсЙЉСЫЫќЖдздМКдЄВтжЪСПЕФКтСПЃЉЁЃШЛКѓЫћУЧЪЙгУетаЉЗжзщРДЪЖБ№ЕААзжЪгюжцЕФвЛаЉзюКкАЕЕФНЧТфЁЃ
вЛИіСюШЫОЊЯВЕФЪЧДгЮДМћЙ§ЕФвЛжжЕААзжЪаЮзДЁЃбаОПШЫдБГЦжЎЮЊЁЎBetaЛЈЁЏЃЌвђЮЊетжжНсЙЙКЌгааэЖрЗЂМазДзЊЭфЁЊЁЊетаЉдкБЛГЦЮЊBetaЭАЕФвбжЊЕААзжЪаЮзДжаПЩвдевЕНЁЊЁЊРрЫЦгкЛЈАъЁЃКЌгаBetaЛЈЕФЕААзжЪБЫДЫЯрОрЩѕдЖЃЌЕЋФПЧАЛЙВЛЧхГўЫќУЧЕФзїгУЃЌе§дкНјвЛВНбаОПИУаЮзДЕФХхРзРЫЕЁЃ
ЁАетЯюЙЄзїЪЕМЪЩЯДђПЊСЫвЛИіХЫЖрРКазгАуЕФЯюФПЁЃЮвУЧБиаыОіЖЈФФаЉжЕЕУгХЯШПМТЧЁЃЁБХхРзРВЙГфЕРЁЃЫ§КЭЭЌЪТЯЃЭћЦфЫћбаОПШЫдБЪЙгУЫћУЧЕФЭјТчРДВщПДЫћУЧзюЯВЛЖЕФЕААзжЪШчКЮШкШыИќЙуЗКЕФЗжзггюжцЁЃ
ТзЖиДѓбЇбЇдКЕФМЦЫуЩњЮябЇМвПЫРяЫЙЭЁЁЄАТТзИъЖдЬНЫїЕААзжЪгюжцЕФаТЗНЗЈИаЕНаЫЗмЁЃЕЋЫ§ОЏИцЫЕЃЌвЛаЉБЛШЯЮЊЖдећИіЕААзжЪИпЖШзМШЗЕФAlphaFoldдЄВтПЩФмВЛЛсзМШЗЕиДњБэбаОПШЫдБИааЫШЄЕФЕААзжЪЕФЙІФмВПЗжЛђгђЕФаЮзДЁЃЦВПЊФЧаЉвьГЃЃЌгІИУШдШЛШУбаОПШЫдБгЕгавЛДѓЖбаТЕФЕААзжЪМвзхЃЌАТТзИъЫЕ ЃЌЁАетЬЋСюШЫаЫЗмСЫЁБЁЃ
СьЕМAlphaFoldЭХЖгЕФЙШИшDeepMindЕФдМКВЁЄжьВЎУЛгаВЮгыетСНЯюбаОПЃЌЫћЖдбаОПШЫдБПЊЗЂаТЗНЗЈРДЬНЫїЫћКЭЫћЕФЭЌЪТУЧЪЭЗХЕФгюжцИаЕНаЫЗмЁЃЫћШЯЮЊетаЉбаОПЪЧвЛИіаТСьгђЕФПЊЪМЃЌдкетИіСьгђжаЃЌЕААзжЪНсЙЙвдФбвдЯыЯѓЕФЙцФЃБЛбаОПЁЃ ЁАЮвЦкД§зХИќЖрЕФЗЂЯжЁЃЁБ
ЁЎA PandoraЁЏs boxЁЏ: map of protein-structure families delights scientists (nature.com)
Clustering-predicted structures at the scale of the known protein universe | Nature
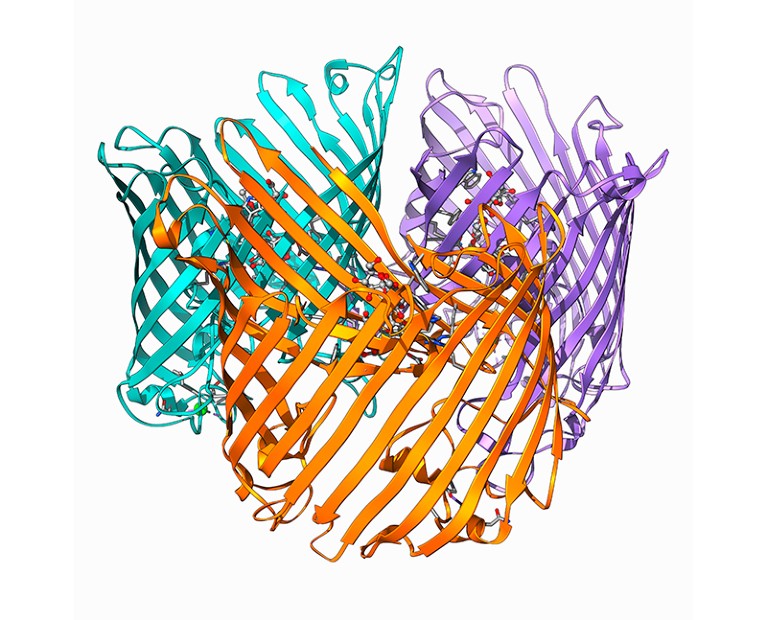
ЕААзжЪЪЧЫљгаЯИАћЙ§ГЬЕФжївЊВЮгыепЃЌДгВњЩњФмСПЕНЯИАћЗжСбЁЃСЫНтЫќУЧЕФНсЙЙЖдгкбаОПЦфЙІФмЁЂНјЛЏвдМАПЩФмЕФвЉЮяЩшМЦЖМжСЙиживЊЁЃОЁЙмЮвУЧЙ§ШЅМИФъЖдЕААзжЪађСаЕФжЊЪЖдіГЄЯрЕБПЩЙлЃЌДяЕНСЫЪ§вкИіађСаЃЌЕЋгЩгкШБЗІИпЖШПЩРЉеЙЕФЪЕбщЗНЗЈЃЌЖдЫќУЧ3DНсЙЙЕФжЊЪЖвЛжБжЭКѓЁЃЯждкЃЌЭЈЙ§ађСадЄВтНсЙЙЕФЗНЗЈ1ЁЂ3ЁЂ4ЕФНјВНЃЌЪЙЕУвбжЊЕААзжЪгюжцЕФЕААзжЪНсЙЙЕФПЩРЉеЙдЄВтГЩЮЊПЩФмЁЃAlphaFoldЕААзжЪНсЙЙЪ§ОнПтЃЈAFDBЃЉЪЧвЛИіЙЋПЊПЩгУЕФЕААзжЪНсЙЙКЭжУаХЖШжИБъЕФЪ§ОнДцДЂПтЃЌЪЙгУAlphaFold2 AIЯЕЭГНјаадЄВтЁЃОЁЙмШдВЛШчЪЕбщШЗЖЈЕФНсЙЙЃЌЕЋдкПМТЧдЄВтОжВПОрРыВювьВтЪдЃЈpLDDTЃЉжУаХЖШжИБъЕФЧщПіЯТЃЌAlphaFoldдЄВтЕФНсЙЙзмЬхЦРЙРЮЊИпжЪСПЁЃAlphaFold2МАЦфдЄВтЕФНсЙЙЯжвбгУгкИїжжгІгУЃЌАќРЈбаОПЕААзжЪПкДќЁЂдЄВтИДКЯЮяНсЙЙбаОПНсЙЙЯрЫЦадаТЕФелЕўдЄВтвдМАЩѕжСИФЩЦЛљвђзщзЂЪЭЁЃ
дЄВтЕААзжЪНсЙЙЕФДѓСПдіМгДйЪЙПЊЗЂИќИпаЇЕФМЦЫуЗНЗЈЃЌАќРЈНсЙЙЪ§ОнЮФМўбЙЫѕПкДќдЄВтЗНЗЈвдМАЭЈЙ§НсЙЙБШЖдБШНЯЕААзжЪНсЙЙЁЃЮЊДЫЃЌвбПЊЗЂСЫFoldseekЁЃгыЯШЧАЕФЗНЗЈЯрБШЃЌFoldseekПЩвдЭЈЙ§НЋНсЙЙЕФБШНЯЫйЖШЬсИпЫФЕНЮхБЖРДдіМгНсЙЙЕФБШНЯЫйЖШЃЌЭЌЪББЃГжУєИаадЃЌЪЙЕУдкДѓЙцФЃЩЯНјааНсЙЙБШНЯГЩЮЊПЩФмЁЃАДНсЙЙЖдЕААзжЪНјааОлРрЪЧЗжЮіНсЙЙЪ§ОнПтЕФЙиМќЙЄОпЃЌвђЮЊЫќПЩвдЪЕЯждЖГЬЯрЙиЕААзжЪЕФЗжзщЁЃЪЖБ№дЖОрРыЙиЯЕПЩФмЮЊСЫНтЕААзжЪНсЙЙбнЛЏКЭЙІФмЬсЙЉгаМлжЕЕФМћНтЁЃР§ШчЃЌдкКИЧШЫРрКЭ20ИіФЃаЭЩњЮяЬхЕФЕААззщжаЃЌГѕЪМЗЂВМЕФдМ36.5ЭђИіНсЙЙЕФЕААзжЪМвзхЗжЮіБэУїЃЌИУМЏКЯжаЕФ92%ЕФдЄВтНсЙЙгђгыЯжгаЕФГЌМвзхЦЅХфЁЃШЛЖјЃЌЪЙгУЕБЧАЗНЗЈНЋЫљга2.14вкИіНсЙЙЯрЛЅБШНЯашвЊДѓдМ10ФъЕФЪБМфЃЌМДЪЙдк64КЫЛњЦїЩЯвВЪЧШчДЫЁЃЮЊСЫМгПьАБЛљЫсађСаЕФОлРрЙ§ГЬЃЌвбОЬсГіСЫвЛжжЯпадЪБМфЫуЗЈLinclust17РДЯджјМѕЩйМЦЫуЪБМфЁЃШЛЖјЃЌетаЉЗНЗЈЩаЮДгІгУгкЛљгкЕААзжЪНсЙЙЯрЫЦадЕФОлРрЁЃ
БОбаОПЗжЮіСЫАќКЌ21.4вкИіПчдНЩњУќжЎЪїЕФдЄВтНсЙЙЕФAlphaFoldЕААзжЪНсЙЙЪ§ОнПтЁЃЮЊСЫФмЙЛЬНЫїетвЛзЪдДЃЌЮвУЧПЊЗЂСЫвЛжжИпЖШПЩРЉеЙЕФНсЙЙЛљОлРрЫуЗЈЃЌЛљгкLinclust17ЃЌдк5ЬьФкЪЙгУ64КЫдк5200ЭђИіНсЙЙЩЯНјааНсЙЙадЖдЦыКЭОлРрЁЃНЋAlphaFoldНсЙЙЪ§ОнПтОлРрГЩ230ЭђИіДиЃЌЦфжа31%ЕФДиДњБэ4%ЕФЕААзжЪађСаЃЌгыЯШЧАвбжЊЕФНсЙЙЛђМвзхзЂЪЭВЛЦЅХфЁЃНсЙћЗЂЯжга532,478ИіДиЕФДњБэДцдкгкећИіЩњУќжЎЪїЩЯЃЌЛЙЗЂЯжСЫМИИіЬиЖЈгкЮяжжЕФНсЙЙДиЃЌПЩФмАќКЌаТЛљвђЕЎЩњЪТМўЕФЪОР§ЁЃзюКѓЃЌЪЙгУНсЙЙБШНЯРДдЄВтМвзхКЭЫќУЧЕФЙиЯЕЃЌЪЖБ№МйЩшЕФдЖГЬЭЌдДЙиЯЕЃЌРЉДѓвбжЊМвзхЕФНјЛЏИВИЧЗЖЮЇЁЃ
https://blog.sciencenet.cn/blog-41174-1402628.html
ЩЯвЛЦЊЃКХжШЫдНРДдНЖрЃЌИљдДЕНЕзЪЧЪВУДЃП
ЯТвЛЦЊЃКШЫЙЄзгЙЌММЪѕНЋНјШыСйДВЪдбщНзЖЮ
