博文
SSB | 清华大学卢元/陈永湘课题组:磷酸化蛋白质的定制化和稳定化合成
|
蛋白质磷酸化在调节细胞生长、代谢、凋亡和信号转导等具有关键作用,与人类疾病息息相关。因此磷酸化蛋白质的合成有助于相关基础和应用研究,然而其合成的关键挑战在于蛋白质磷酸化修饰的精准性和稳定性。清华大学卢元课题组和陈永湘课题组合作,发展了基于非天然磷酸化氨基酸嵌入的无细胞蛋白质高效合成和筛选系统,利用其开放性、毒性耐受性、灵活调控等优势,通过底盘选择、非天然正交系统筛选等,成功实现蛋白质的高效精准磷酸化;并进一步人工合成更稳定不易水解的非天然磷酸化氨基酸,将其成功嵌入功能蛋白质,解决磷酸化蛋白质的稳定性问题。该工作为开发高度可扩展的无细胞非天然蛋白质合成平台和用户自定义磷酸化蛋白质的高效生产奠定了基础,并将大大促进对蛋白质磷酸化修饰机制和重大疾病诊治药物开发的研究。相关工作以“Customized synthesis of phosphoprotein bearing phosphoserine or its nonhydrolyzable analog”为题,发表在Synthetic and Systems Biotechnology期刊上。
研究内容简介 Studies on the mechanism of protein phosphorylation and therapeutic interventions of its related molecular processes are limited by the difficulty in the production of purpose-built phosphoproteins harboring site-specific phosphorylated amino acids or their nonhydrolyzable analogs. Here we address this limitation by customizing the cell-free protein synthesis (CFPS) machinery via chassis strain selection and orthogonal translation system (OTS) reconfiguration screening. The suited chassis strains and reconfigured OTS combinations with high orthogonality were consequently picked out for individualized phosphoprotein synthesis. Specifically, we synthesized the sfGFP protein and MEK1 protein with site-specific phosphoserine (O-pSer) or its nonhydrolyzable analog, 2-amino-4-phosphonobutyric acid (C-pSer). This study successfully realized building cell-free systems for site-specific incorporation of phosphonate mimics into the target protein. Our work lays the foundation for developing a highly expansible CFPS platform and the streamlined production of user-defined phosphoproteins, which can facilitate research on the physiological mechanism and potential interference tools toward protein phosphorylation. Fig. 1. Flexible CFPS platform with chassis strain selection and OTS reconfiguration screening for the customized synthesis of phosphoprotein. (a) Limitations of site-specific pSer incorporation by the genetic code expansion (GCE) strategy: poor cell permeability and OTS orthogonality, as well as competition for the target UAG stop codon. (b) Schematic of the process of cell-free protein expression employing crude extracts from the reengineered chassis strains. (c) Precise control of OTS concentration and addition methods on protein expression conditions for the streamlined production of user-defined phosphoproteins. (d) Flexible chassis strains reengineering and chassis strain selection. (e) High-throughput OTS reconfiguration screening in the setting of selected chassis strains. Fig. 2. Chassis strain selection with endogenously expressed OTS. (a) Design of genetically modified chassis strains with endogenous OTSs system. One-plasmid system included Sep-RS, EF-Sep and Sep-tRNA in the EcAR7. ΔA, C321. ΔAΔSerB and BL21ΔSerB strains. (b) Preparation of endogenous OTSs. The cell extracts containing endogenous OTS system were from genetically recoded strains with one-plasmid OTS system overexpression. (c) The structures of O-pSer and C-pSer. (d–f) The results of Phospho-sfGFP expression during chassis strains selection with endogenous OTSs system in C321. ΔAΔSerB (d), EcAR7. ΔA (e) and BL21. ΔSerB (f) strains. TAG codon at position 2 or 23 directed pSer incorporation into sfGFP, which were named 2TAG-sfGFP or 23TAG-sfGFP. The sfGFP expression was catalyzed by extracts derived from genetically recoded strains, C321. ΔAΔSerB (d), EcAR7. ΔA (e) and BL21. ΔSerB (f). The expression level was determined by fluorescence intensity of generated sfGFP protein. When the relative fluorescence was 1, the corresponding protein expression levels were 1.25 mg/mL (d), 0.58 mg/mL (e), and 2.55 mg/mL (f), respectively. Error bars reported SD from three biological replicates.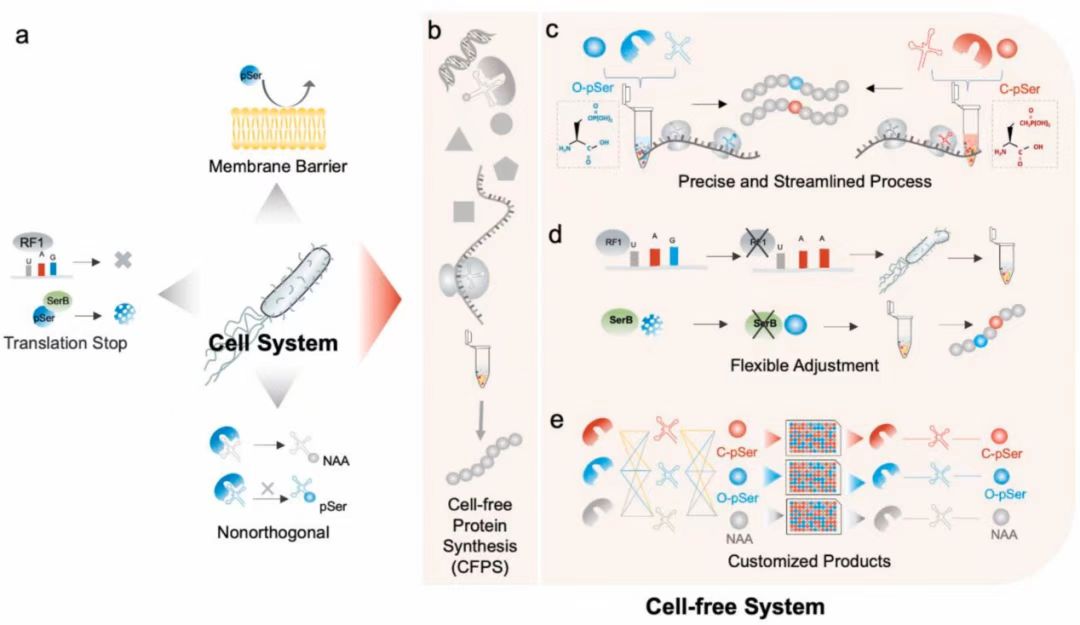

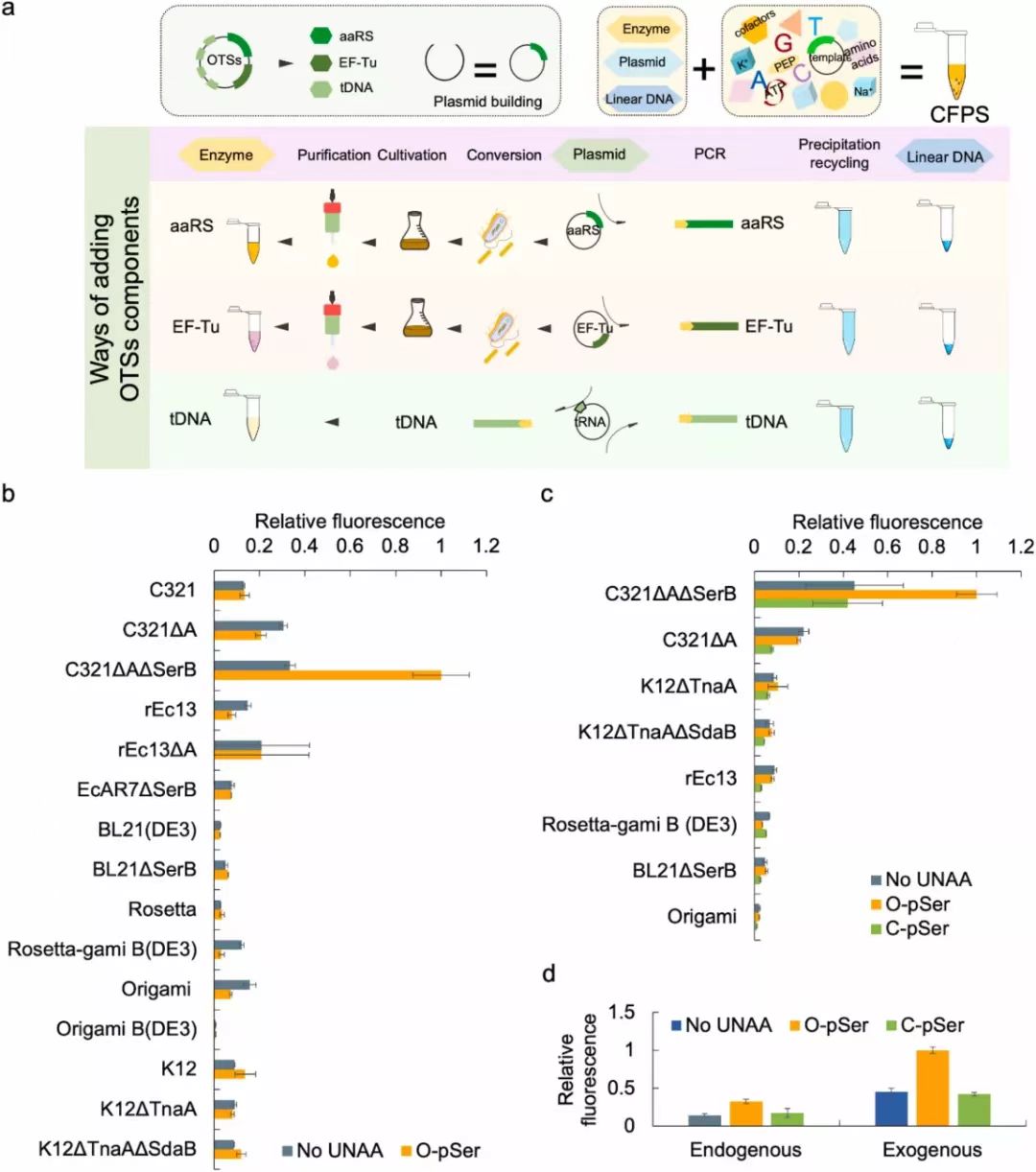
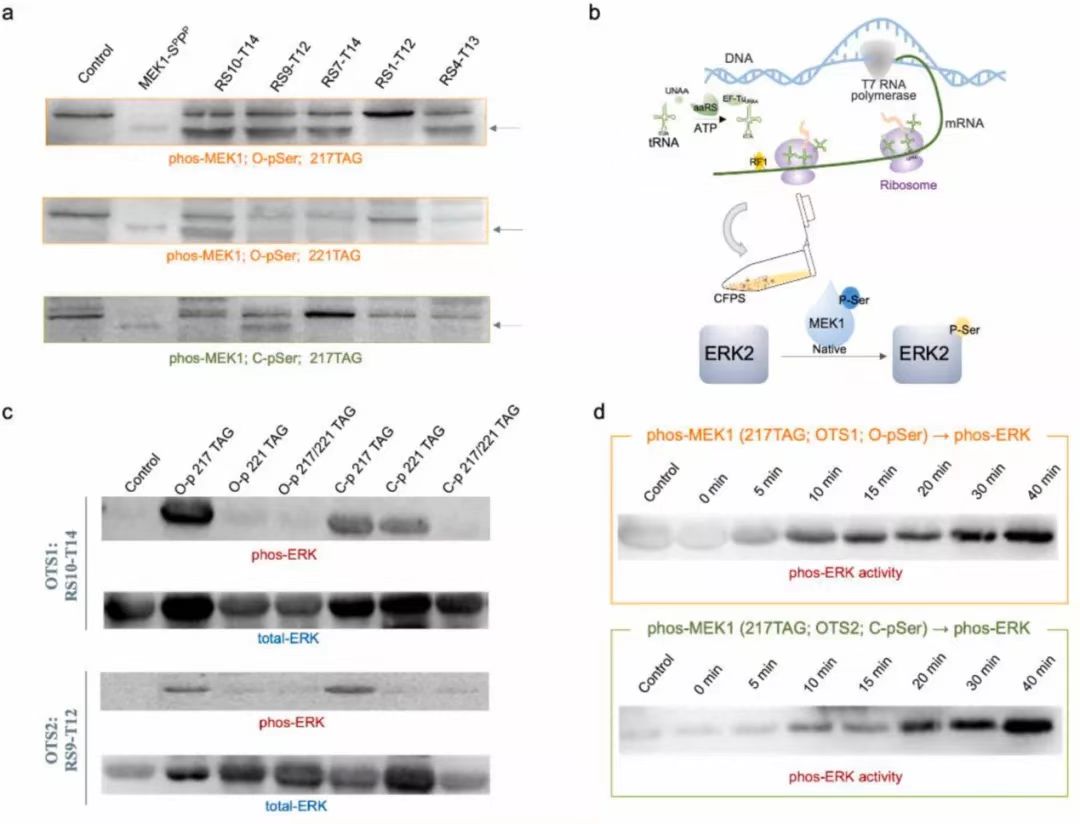
Fig. 3. Chassis strain selection with exogenously added or expressed OTS. (a) Preparation and purification of OTSs components. (b) The results of O-Phospho-sfGFP expression during chassis strains selection with exogenous OTSs. The cell-free reactions were supplemented with the necessary cell extracts derived from 16 kinds of genetically recoded chassis strains (Supplementary Table 1). TAG codon at position 23 directed O-pSer incorporation into sfGFP, and as a control with no UNAA added. The expression level was determined by fluorescence intensity of generated sfGFP protein. Error bars report SD from three biological replicates. Extended data with a TAG codon at position 2 was shown in Supplementary Fig. S5. (c) The results of O-Phospho-sfGFP and C-Phospho-sfGFP expression, using cell extracts derived from 8 kinds of genetically recoded chassis strains with top performance on O-pSer incorporation. TAG codon at position 23 directed O-pSer and C-pSer incorporation into sfGFP, and as a control with no UNAA added. The expression level was determined by fluorescence intensity of generated sfGFP protein. Error bars report SD from three biological replicates. Extended data with a TAG codon at position 2 was shown in Supplementary Fig. S6. (d) The comparison of sfGFP expression for chassis strains selection with endogenous and exogenous OTSs, respectively. Error bars report SD from three biological replicates. Fig. 4. High-throughput screening of exogenous OTSs reconfiguration orthogonality. (a) (b) The orthogonality analysis for reconfigured OTS components (aaRS and tRNA for O-pSer or C-pSer incorporation). The heatmap depicted the fluorescence value ratio of the expressed sfGFP (UNAA-added CFPS reaction) to the sfGFP expression baseline (non UNAA-added CFPS reaction). The cell-free reactions were supplemented with the necessary cell extracts derived from genetically recoded C321. ΔAΔSerB strain. A TAG codon at position 23 directed O-pSer (a) and C-pSer (b) incorporation into sfGFP. (c) Structure and sequence analysis of RS0-T13 pair for top-performing O-pSer incorporation. (d) Structure and sequence analysis of RS4-T1 pair for top-performing C-pSer incorporation. The tRNA secondary structure was predicted by RNAfold webserver, and MFE structure drawing encoding base-pair probabilities was shown. Fig. 5. Cell-free synthesis and activity assessment of site-specifically phosphorylated MEK1. (a) Quantitation analysis of site-specifically phosphorylated MEK1 production by western blot. (b) Schematic of the process of phosphorylated MEK1 production via cell-free reaction and the subsequent in vitro kinase assay using the native MEK1 substrate ERK2. (c) In vitro MEK1 kinase activity was assayed using selected OTS reconfiguration and using ERK2 as a substrate. The far-left lane was background ERK2 phosphorylation without MEK1 protein addition. Total ERK was determined by western blot analysis as a control. (d) The time points of in vitro MEK1 kinase activity were measured at 0, 5, 10, 15, 20, 30, and 40 min.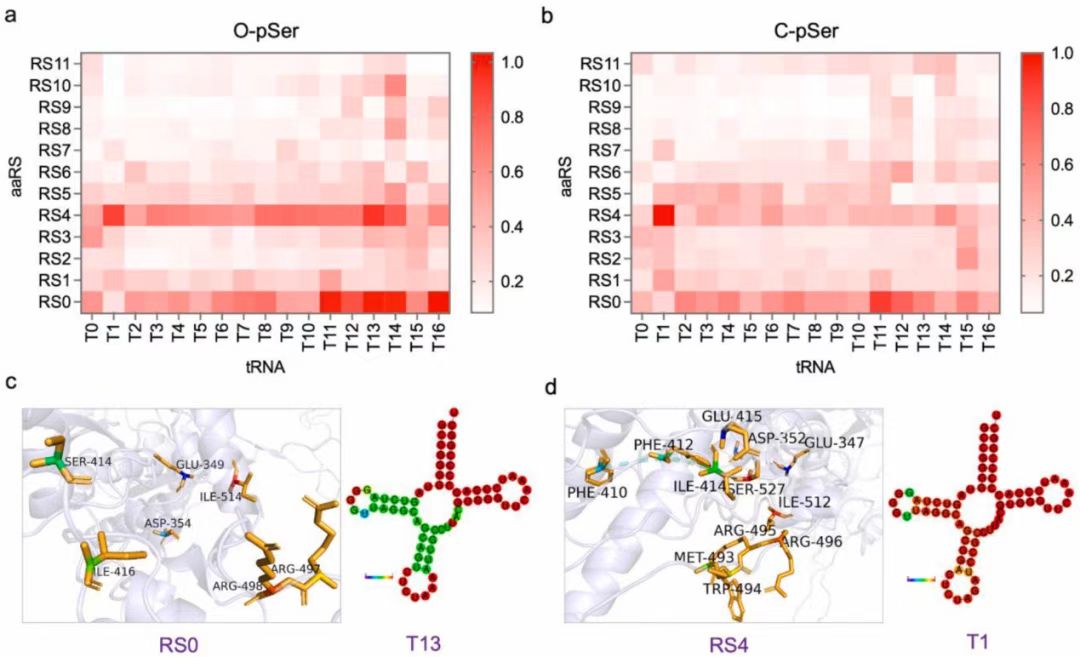
Customized synthesis of phosphoprotein bearing phosphoserine or its nonhydrolyzable analog Dong Liu, Yingying Liu, Hua-Zhen Duan, Xinjie Chen, Yanan Wang, Ting Wang, Qing Yu, Yong-Xiang Chen, Yuan Lu. https://doi.org/10.1016/j.synbio.2022.11.004 2021 Impact Factor: 4.692; 5-Year Impact Factor: 5.23; JCR分区Q2 2021 CiteScore: 6.60, 位列学科Q1区 2022中科院分区生物学大类Q2区;生物工程与应用微生物小类Q1区 入选2019年中国科技期刊卓越行动计划高起点新刊项目
https://blog.sciencenet.cn/blog-3496796-1380187.html
上一篇:SSB | 华东理工大学和上海交通大学开发基于硫修饰DNA的新型快速核酸检测平台
下一篇:SSB | 华东理工大学和上海交通大学开发基于硫修饰DNA的新型快速核酸检测平台
