博文
《免疫学前沿》:免疫细胞有后备机制
||
《免疫学前沿》:免疫细胞有后备机制
诸平
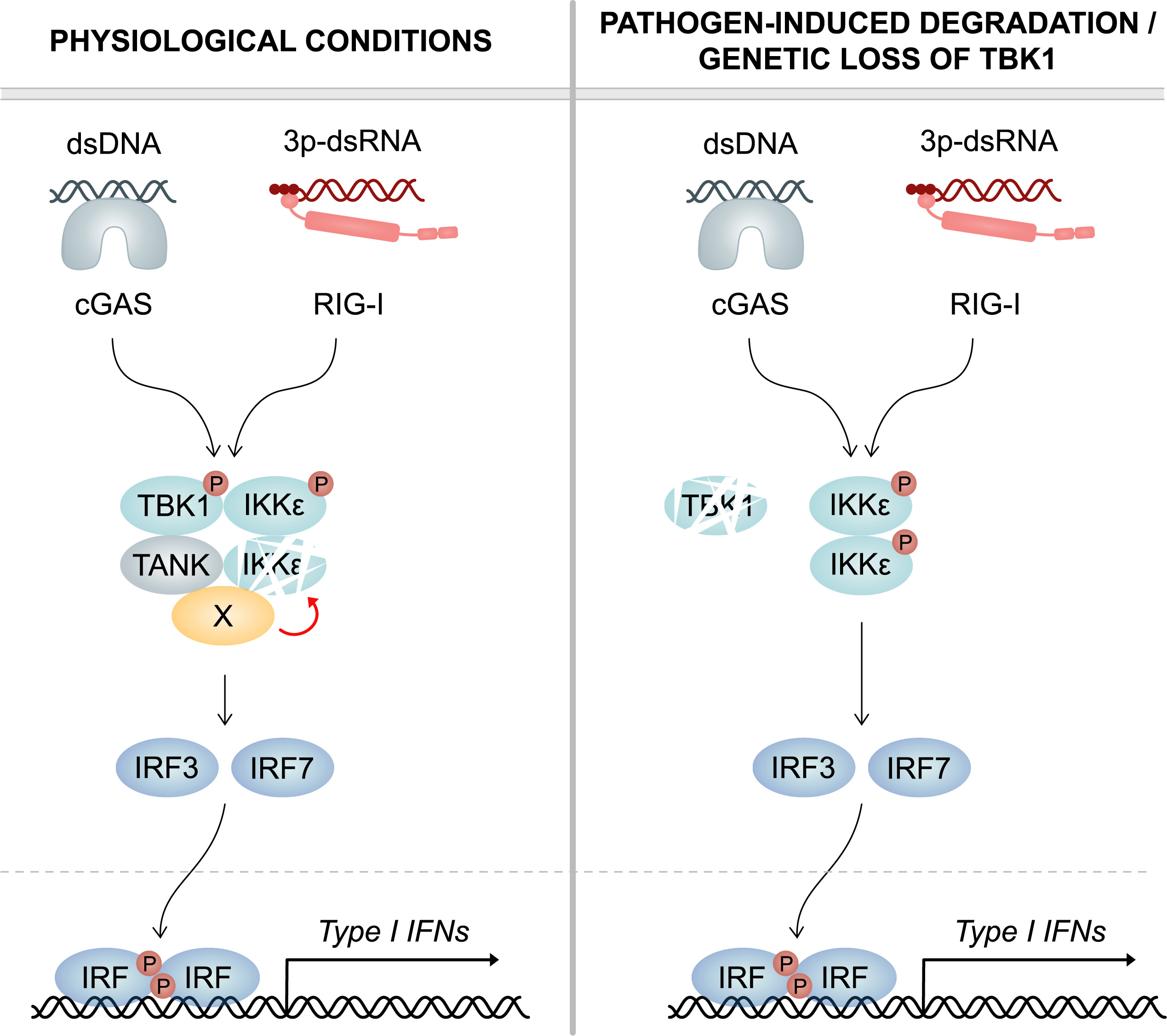
Left: Under physiological conditions, - TBK1 continuously induces the degradation of its cognate kinase IKKepsilon. For this purpose, TBK1 recruits an as yet unidentified cofactor (X) via its scaffold protein TANK, which destabilizes IKKepsilon. Upon infection, both TBK1 and IKKepsilon contribute to the production of interferon (type I IFN). Right: Upon pathogen-induced degradation or genetic loss of TBK1, it no longer reduces IKKepsilon protein stability. Thus, IKKepsilon protein levels are greatly enhanced, which compensates for the deficiency of TBK1. This ensures an unrestrained type I IFN response and efficient clearance of infection.© Figure: Frontiers in Immunology/DOI: 10.3389/fimmu.2023.1073608
据德国波恩大学(University of Bonn / Universität Bonn)2023年3月10日报道,波恩的科学家研究发现免疫细胞有后备机制(Immune cells have a backup mechanism)。
如果TBK1酶(If the TBK1 enzyme)发生突变,病毒感染的易感性就会增加——除非它缺失(If the TBK1 enzyme is mutated, susceptibility to viral infections increases – unless it is missing)
TBK1酶是先天免疫系统的重要组成部分,在抵御病毒方面发挥着关键作用。在突变诱导的 TBK1功能丧失后,患者表现出对病毒感染的易感性增加。引人注目的是,如果TBK1根本不表达,则看不到这种临床效果。波恩大学医院(University Hospital Bonn)和波恩大学卓越免疫感觉2集群(Cluster of Excellence Immuno Sensation 2 at the University of Bonn)的马丁·舒立(Martin Schlee)教授领导的研究团队,现在已经阐明了这种假设差异背后的机制。相关研究结果于2023年3月3日已经在《免疫学前沿》(Frontiers in Immunology)杂志网站发表——Wegner Julia, Hunkler Charlotte, Ciupka Katrin, Hartmann Gunther, Schlee Martin. Increased IKKepsilon protein stability ensures efficient type I interferon responses in conditions of TBK1 deficiency. Frontiers in Immunology(Sec. Molecular Innate Immunity), 03 March 2023. Vol. 14. DOI: 10.3389/fimmu.2023.1073608. https://doi.org/10.3389/fimmu.2023.1073608
上述图示中左图:在生理条件下,TBK1持续诱导其同源激酶 IKKepsilon(cognate kinase IKKε) 的降解。为此,TBK1通过其支架蛋白TANK招募了一种尚未确定的辅助因子 (X),这会破坏IKKε的稳定性。感染后,TBK1和IKKε都有助于产生干扰素(I 型 IFN)。右图:在病原体诱导的TBK1降解或遗传丢失后,它不再降低IKKε蛋白的稳定性。因此,IKKε蛋白水平大大提高,从而弥补了TBK1的不足。这确保了不受限制的I型干扰素反应和有效的感染清除。
在人体中,病毒颗粒是由位于细胞内或细胞表面的所谓模式识别受体(pattern recognition receptors简称PRR) 来识别。激活后,信号级联被初始化,最终导致信号分子如干扰素(interferons)和细胞因子(cytokines)的产生和释放。这些信使分子提醒邻近的免疫细胞并指出病毒感染,从而引发免疫反应。
该信号级联的一部分是TANK结合激酶1 (TANK Binding Kinase 1简称TBK1)。如果PRR检测到病毒颗粒,则TBK1会被激活。TBK1反过来激活两个转录因子,它们进入细胞核,在此它们诱导干扰素和细胞因子基因的转录。
对病毒感染的易感性(Susceptibility to viral infections)
TBK1 基因的点突变(Point-mutations)可能导致TBK1功能丧失。在人类中,这表现为对病毒感染的临床易感性。引人注目的是,如果TBK1不表达并且在细胞中完全缺失,则不会观察到这种效果。“令人惊讶的是,TBK1在人类中的完全缺失与抗病毒反应的降低无关,”波恩大学医院临床化学和临床药理学研究所(Institute of Clinical Chemistry and Clinical Pharmacology at the University Hospital Bonn)的马丁·舒立教授说。到目前为止,还不清楚为什么TBK1表达完全缺失在免疫能力方面比TBK1突变影响激酶功能更耐受。
波恩研究人员现在已经能够为这些以前无法解释的观察结果提供解释。“与TBK1非常相似的第二种酶在其中起着重要作用:IkB激酶ε(IkB kinase epsilon),简称 IKKε(IKKepsilon),”该研究的第一作者朱莉娅· 韦格纳(Julia Wegner)博士解释道。就像TBK1一样,IKKε作用于PRR的下游并控制干扰素的表达。这两种蛋白质在结构上也非常相似,序列同源性超过60%。一项新发现是TBK1对IKKε有直接影响。“在骨髓细胞中,我们可以证明TBK1 调节相关激酶 IKKε的表达,”朱莉娅· 韦格纳博士补充道。
毫无办法(No half measures)
TBK1降低了IKKε的稳定性。这个过程与蛋白质的酶功能无关。临床化学和临床药理学研究所(Institute of Clinical Chemistry and Clinical Pharmacology)所长兼免疫感觉2卓越集群(Immuno Sensation 2 Cluster of Excellence)发言人巩特尔·哈特曼(Gunther Hartmann)教授解释说:“因此,由于点突变而失去功能的TBK1仍然能够破坏IKKε的稳定性。这导致人类免疫细胞中激酶 IKKε的持续降解。”
因此,TBK1表达的缺失导致IKKε的丰度增加。这种机制确保即使没有TBK1也能发生抗病毒免疫反应。另一方面,由点突变引起的TBK1功能丧失并不能阻止IKKε的不稳定和降解,因此最终这两个因素都无法用于病毒防御。结果是对病毒感染的易感性增加。
病毒的武器(Weapons of a virus)
因此,在健康的生物体中,增加IKKε的量可以补偿TBK1的损失。当病毒专门寻求消除人体自身的防御时,这一点就变得尤为重要。单纯疱疹病毒1(Herpes simplex virus 1简称HSV-1)、人类免疫缺陷病毒 (human immunodeficiency virus简称HIV) 以及严重急性呼吸系统综合症冠状病毒2 (severe acute respiratory syndrome coronavirus 2简称SARS-CoV-2) 都能够特异性诱导TBK1降解。此外,一些细菌种类能够引起TBK1的降解。朱莉娅· 韦格纳博士解释说:“我们的数据清楚地表明,人类免疫细胞具有重要的后备机制。即使发生病原体诱导的TBK1降解,它们也能够维持有效的抗病毒反应。此外,该机制在TBK1遗传丢失的情况下也会起作用。”
本研究得到了德国研究基金会(Deutsche Forschungsgemeinschaft简称DFG)在德国卓越战略下资助(DFG under Germany’s Excellence Strategy – EXC2151 – 390873048)、其它项目(DFG Project ID 369799452 TRR237, Project ID 397484323 TRR259, DFG SCHL1930/1-2)以及德国传染病中心(German Center for Infectious Diseases简称DZIF TTU 07.834_00)的支持或资助。
上述介绍,仅供参考。欲了解更多信息,敬请注意浏览原文或者相关报道。
TBK1 and IKKε are related, crucial kinases in antiviral immune signaling pathways downstream of cytosolic nucleic acid receptors such as cGAS and RIG-I-like receptors. Upon activation, they phosphorylate the transcription factors IRF3 and IRF7 and thereby initiate the expression of type I interferons and antiviral effectors. While point mutation-induced loss of TBK1 kinase activity results in clinical hyper-susceptibility to viral infections, a complete lack of TBK1 expression in humans is unexpectedly not associated with diminished antiviral responses. Here, we provide a mechanistic explanation for these so-far unexplained observations by showing that TBK1 controls the protein expression of its related kinase IKKε in human myeloid cells. Mechanistically, TBK1 constitutively diminishes the protein stability of IKKε independent of TBK1 kinase activity but dependent on its interaction with the scaffold protein TANK. In consequence, depletion of TBK1 protein but not mutation-induced kinase deficiency induces the upregulation of IKKε. Due to the functional redundancy of the kinases in cGAS-STING and RIG-I-like receptor signaling in human myeloid cells, enhanced IKKε expression can compensate for the loss of TBK1. We show that IKKε upregulation is crucial to ensure unmitigated type I interferon production in conditions of TBK1 deficiency: While the type I interferon response to Listeria monocytogenes infection is maintained upon TBK1 loss, it is strongly diminished in cells harboring a kinase-deficient TBK1 variant, in which IKKε is not upregulated. Many pathogens induce TBK1 degradation, suggesting that loss of TBK1-mediated destabilization of IKKε is a critical backup mechanism to prevent diminished interferon responses upon TBK1 depletion.
https://blog.sciencenet.cn/blog-212210-1380281.html
上一篇:科学家们发现了一种可能引发地球生命的物质
下一篇:“出乎意料”——研究人员查明了太阳“类心跳”信号的神秘来源