博文
元素周期表能够与电子组态“更完美地结合”吗?
 精选
精选
|||
元素周期表能够与电子组态“更完美地结合”吗?
2019年,元素周期表诞生150周年了。联合国教科文组织宣布2019年为“元素周期表年”。150年来,元素周期表在化学理论和实践中都发挥了巨大的作用。我们纪念元素周期表150周年是一件很有意义的事情。
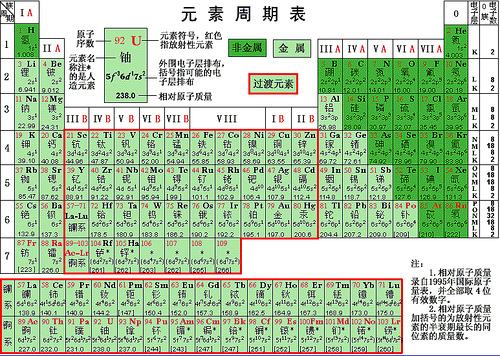
众所周知,元素周期表是人们根据当时已知的元素的物理和化学性质的周期性变化而提出来的。这是一个在150年前的天才创造,当时人们并不知道原子是什么,不知道它的组成,更不知道它的结构。在人们搞清楚了原子的组成和结构之后,人们发现,元素周期表之所以有这样的形式,是由于原子的结构也就是原子中的电子在其轨道上的排布所决定的。元素性质的周期性变化,只是其原子结构也就是其电子所占据的轨道,特别是最外层占据情况的周期性变化的反映。
早在20世纪30年代,对于电子在轨道上被占据的先后顺序,就有所谓马德隆规则。根据此规则,原子轨道能级的高低由轨道的主量子数n与角动量量子数l(l为最小0,最大n-1的整数)共同决定:n+l值较大者其能级较高;若n+l值相等,则n大者能级高。依此,原子中的电子所占有的轨道从能量最低的轨道开始自下往上排布。
马德隆规则对于能级由低至高的次序为:
1s<2s<2p<3s<3p<4s<3d<4p<5s<4d<5p<6s<4f<5d<6p……
这个规则,与我们现在的元素周期表里各化学元素的电子组态的排列次序是基本一致的。
说基本一致,那就是还是有不一致的地方。例如,第24号元素铬,根据上述规则,其最外层电子组态似乎应当是4s23d4,也就是说,有2个电子在4s轨道,4个电子在3d轨道,但是,实际上根据光谱得到的电子组态似乎应当是4s13d5,即只有1个电子在4s轨道,5个电子在3d轨道。大概有20来种元素的电子组态与马德隆规则有略不相符的电子组态。
那么是不是能够找到这些原子的电子组态“出轨”的规律呢?似乎也没有一个很好的统一的规律。
于是,有一些人对此不是很满意,他们总是企图想办法弥补这个缺点,以使得元素周期表能够显得更加完美。但是,他们的努力似乎都很难有什么用,就像一条不够长的被子要盖上一个大个子的身体似的,顾了头就顾不了脚,顾了脚又顾不了头。
那么,难道就不能使得元素周期表比现在更加“完美”一点,找到一个解决上面所说问题的办法吗?
为了找到问题的症结,人们需要“追根寻源”。
我们的问题是什么?是原子的电子组态与周期表的“列”并不完全吻合。
什么是原子的电子组态?就是该原子的电子在原子轨道上的占有情况。
什么是原子轨道?原子轨道的概念是从哪里来的?解薛定谔方程所得到的解。
继续追问:求解的是什么体系的薛定谔方程?求解的是氢原子体系的薛定谔方程。求解的时候,由于边界条件的限制,引出了三个量子数:主量子数n,角动量量子数l、磁量子数m,以及体系能量的分立值。氢原子体系的薛定谔方程由n、l、m所确定的体系的状态(波函数)就称为氢原子的原子轨道。沿用光谱学的符号,把角量子数l的值为0、1、2、3…的轨道分别表示为s、p、d、f…。这样,我们就知道了,这些1s2s2p3s3p4s3d4p等等轨道都是氢原子或者类氢离子的薛定谔方程的解,这是一个单电子波函数。
其他原子即多电子原子的薛定谔方程是否也能够严格解出,得到与氢原子类似的原子轨道?多电子原子的薛定谔方程无法像氢原子体系那样严格的解出。
多电子原子的薛定谔方程与氢原子的薛定谔方程的不同在哪里?决定各个体系之间薛定谔方程差别的,在于各体系的哈密顿算符。氢原子的哈密顿算符,有原子核和电子的动能算符,以及包含电子与原子核之间相互吸引的势能。而多电子原子的哈密顿算符中,其势能除了电子与原子核的吸引势能之外,还包含了电子之间的排斥势能。也正因为有这一项电子间的排斥势,使得除氢原子外,多电子原子体系的薛定谔方程,都无法分离变量,从而无法严格求解。因而,所有的多电子原子体系,严格地说,并没有“原子轨道”。
那么,我们现在大家使用的多电子原子的原子轨道,是从哪里来的呢?那是应用近似方法的结果。
对于有n个电子的原子,一个电子受到的作用势,有原子核对该电子的吸引势和其他n-1个电子对它的排斥势。如果人们把这些作用势作平均,也就是假设电子处在原子核和其他n-1个电子的平均势场中,那么这个n电子体系的薛定谔方程就能够分离变量,变成n个类氢离子的薛定谔方程,从而得到类似于氢原子的原子轨道。这就是我们所说的多电子原子的原子轨道。换句话说,多电子原子的原子轨道是在所谓“平均势场”近似下所得到的结果。
由于一个电子所受到的平均势场是与其他n-1个电子的状态(也就是它们的波函数)有关的,所以每一个电子的波函数与其他n-1个电子的波函数有关。人们先假设了这n个单电子波函数,然后用迭代的方法,求得这个体系的各个单电子波函数及其对应的能量,这就是所谓自洽场方法,即Hartree-Fock方法。
一般的Hartree-Fock方法只是考虑了体系处在基组态的情况,把体系的基态波函数写为由基组态的原子轨道组成的Slater行列式。人们把这种近似称为单电子近似。
元素周期表上所标明的各个原子的电子组态,就是它们的基组态。
由于Hartree-Fock方法,只是考虑了平均的势场,所以它对于电子之间的相互作用是考虑不够的,即使把这些原子轨道做最精确的级数展开,体系的能量也只能趋向于某一个极限值,即Hartree-Fock极限。这个极限,离开体系的真实能量还有一个相当大的误差。
拿氦原子来说,这个误差值可以达到1.44%,对于其他多电子原子这个误差值也大多在1%的数量级上,应当说这是一个相当大的误差了。因为这是总能量的误差,而化学反应中体系能量的变化,一般也在总能量1%的数量级上。所以,从理论上说,用Hartree-Fock方法计算到的体系能量对于计算化学反应的能量并没有普遍性的意义。
如果想进一步求解得多电子原子的精确的能量,能够解决化学反应中的能量问题,人们在计算中,就应当考虑“组态相互作用”的问题,也就是说,不能仅仅用上面Hartree-Fock方法,不能认为体系只是处在那个最低的那个电子组态,要考虑基组态与激发组态的相互作用,要把体系的基态波函数写为由基组态行列式和许多个激发组态行列式的线性组合。这种方法,称为组态相互作用方法。
但是,当人们应用组态相互作用方法,即把体系的基态波函数写成由许多个Slater行列式的线性组合时,实际上已经放弃了单电子近似,也就是说,在这种要求得到精确能量的情况下,这些“原子轨道”已经没有任何物理意义了。
总而言之,原子轨道不是一个“物理实在”,或者说,不是一个可以测量的物理量。它只是人们用以解释原子性质而提出来的一个概念。原子轨道这个概念,只是对氢原子这个单电子原子而言基本上是严格的。而对于多电子原子,原子轨道是在“单电子近似”方法下产生的结果。原子轨道这个概念,能够用来解释很多化学现象、解释物质的一些化学和物理性质。
元素的化学和物理性质,包括原子光谱,都是元素本身的性质,是物理测量的结果。而原子轨道和元素的电子组态是人们在一定的近似假定下,用以解释化学和物理性质的一个模型。例如,一根根的光谱线,是人们测量得到的物理实在,而人们用从一个轨道到另一个轨道的跃迁来解释这些谱线,则是一个利用模型进行解释的结果。这个理论模型并不是物理实在,它是对物理实在的近似,具体对这个问题而言,原子轨道是在单电子近似下形成的模型。
对于原子轨道,人们——无论是化学工作者或者光谱工作者,都不可以把它们看得过于“真”,以为是不可撼动的很精确的甚至严格准确的东西。
元素周期表是人们对元素化学和物理性质的总结,原子的电子组态则是单电子近似下的理论模型。周期表所表示的元素的周期性与原子的电子组态的周期性的一致性,是物质宏观性质与微观结构对应关系的反映,是人们认为世界的一大成就。
但是,对于某一个具体的化学元素的性质而言,元素周期表和元素的电子组态都不是精确的。所以,要想让它们完全没有任何“缝隙”的结合实际上是一个不可能完成的任务。
https://blog.sciencenet.cn/blog-612874-1196239.html
上一篇:米洇汤
下一篇:说一所提倡学生自主学习的高中的几件事
全部作者的精选博文
全部作者的其他最新博文
全部精选博文导读
相关博文
- • Challenging Traditional Mathematical Paradigms for AI (初学者版)
- • Yucong Duan\´s Paradox of Mathematics for AI Semantics(初学者版)
- • Paradox of Mathematics by Yucong Duan in DIKWP(初学者版)
- • Traditional Remedy for Traditional Mathematics(初学者版)
- • Traditional Mathematization of DIKWP Semantics(初学者版)
- • MDPI 学者沙龙系列——西安专场圆满落幕