博文
2020-11-30=codon=usage=GB=2019
||
volume 20, Article number: 119 (2019)
Abstract
Background
The uneven use of synonymous codons in the transcriptome regulates the efficiency and fidelity of protein translation rates. Yet, the importance of this codon bias in regulating cell state-specific expression programmes is currently debated. Here, we ask whether different codon usage controls gene expression programmes in self-renewing and differentiating embryonic stem cells.
Results
Using ribosome and transcriptome profiling, we identify distinct codon signatures during human embryonic stem cell differentiation 我们在人类胚胎干细胞分化过程中发现了完全不同的密码子信号. We find that cell state-specific codon bias is determined by the guanine-cytosine (GC) content of differentially expressed genes. 细胞状态特异性的密码子偏好是由差异表达基因的GC含量决定的. By measuring the codon frequencies at the ribosome active sites interacting with transfer RNAs (tRNA), we further discover that self-renewing cells optimize translation of codons that depend on the inosine tRNA modification in the anticodon wobble position. 自我更新细胞的密码子翻译优化取决于在反密码子环摆动位置的肌苷tRNA修饰 Accordingly, inosine levels are highest in human pluripotent embryonic stem cells. 因此,在人多能胚胎干细胞中肌苷水平最高。 This effect is conserved in mice and is independent of the differentiation stimulus.
问题:差异表达基因的GC含量存在差异?是只在细胞分化过程中表现出来还是在各种细胞类型的marker基因中都能够体现?
Conclusions
We show that GC content influences cell state-specific mRNA levels GC含量影响细胞状态特异性mRNA的水平, and we reveal how translational mechanisms based on tRNA modifications change codon usage in embryonic stem cells. 在胚胎干细胞中,翻译机器基于tRNA修饰变化从而改变密码子使用频率
Background
Understanding normal tissue development and disease susceptibility requires the knowledge of mechanisms determining lineage fate decisions in embryonic stem and progenitor cells. While the transcriptional networks governing the naïve and lineage committed states are now increasingly understood [,,], it remains largely unknown whether and how translational mechanisms contribute to early cell fate decisions.
Translation of mRNA takes place on ribosomes, and distinct sets of tRNAs link each nucleotide triplet in mRNA to a corresponding amino acid. The genetic code is degenerate, because the 64 codons (triplets) encode for only 20 amino acids. Thus, single amino acids are often encoded by multiple synonymous codons that are not used randomly. The uneven use of synonymous codons in the transcriptome is commonly found in different organisms and called codon bias []. The selective use of optimal codons that correspond to abundant tRNAs has been suggested to improve translational efficiency by fine tuning translation elongation rates [,,]. Yet, the importance of codon bias in regulating gene expression programmes remains debated, because other features of the coding sequence, such as guanine-cytosine (GC) content and mRNA secondary structure, also strongly influence translation elongation efficiency [, ].
The hundreds of tRNA genes interspersed in the human genome encode only 47 out of the 64 possible tRNA anticodons. Codons lacking a corresponding tRNA are translated via wobble base pairing 通过摆动碱基配对翻译缺少相应tRNA的密码子. While the standard Watson-Crick base pairing is required at the first and second positions, so-called wobbling at the third position can allow otherwise-disfavored base pairing such as between guanosine and uracil [, ]. The affinity by which synonymous codons are recognized via wobble base pairing can vary, and the translation kinetics of different codon-anticodon pairs is further influenced by modified nucleotides present in tRNAs []. Specifically, position 34 in tRNAs, corresponding to the wobble position, carries a wide range of chemical modifications, including 2’O-methylribose, 5-methylcytidine, 5-methoxycarbonylmethyl, 5-carbamoylmethyl, and their derivatives, as well as direct adenine-to-inosine editing (A34I) [,,]. These modifications can dramatically alter codon translation rates and for instance adapt protein synthesis to external stress stimuli [,,,,,,,].
Whether optimal codon usage through tRNA modifications can regulate cell fate has been unexplored. Therefore, using human embryonic stem cells (hESCs) as a model system, we analysed codon usage optimization during self-renewal and differentiation. We triggered the exit of pluripotency by exposure to retinoic acid [, ]. We found that the differentially expressed genes between the cell states were strongly biased by GC content. 我们发现在细胞状态之间差异表达基因存在显著的GC含量偏倚 By analysing the translation kinetics at the ribosomes, we found that self-renewing embryonic stem cells optimized codon usage of specific codons that depended on inosine-modified tRNAs for translation 自我更新的胚胎干细胞优化了依赖于肌苷修饰的tRNA进行翻译的特定密码子的密码子使用. The differential codon bias was independent of the nature of the differentiation stimulus. In summary, we reveal a codon bias driven by tRNA modifications that defines pluripotent embryonic stem cells.
Results
RNA expression and translation highly correlate in self-renewing and differentiating cells
To determine mRNA expression and translation levels in self-renewing and differentiating human embryonic stem cells (hESCs), we performed RNA-sequencing (RNA-seq) and ribosome footprint profiling (Ribo-seq). We induced the exit of pluripotency of hESCs by removing the growth factor FGF-2 and exposing the cells to retinoic acid (RA), one of the best characterized stimuli to induce early embryonic differentiation (Fig. a) [, ].
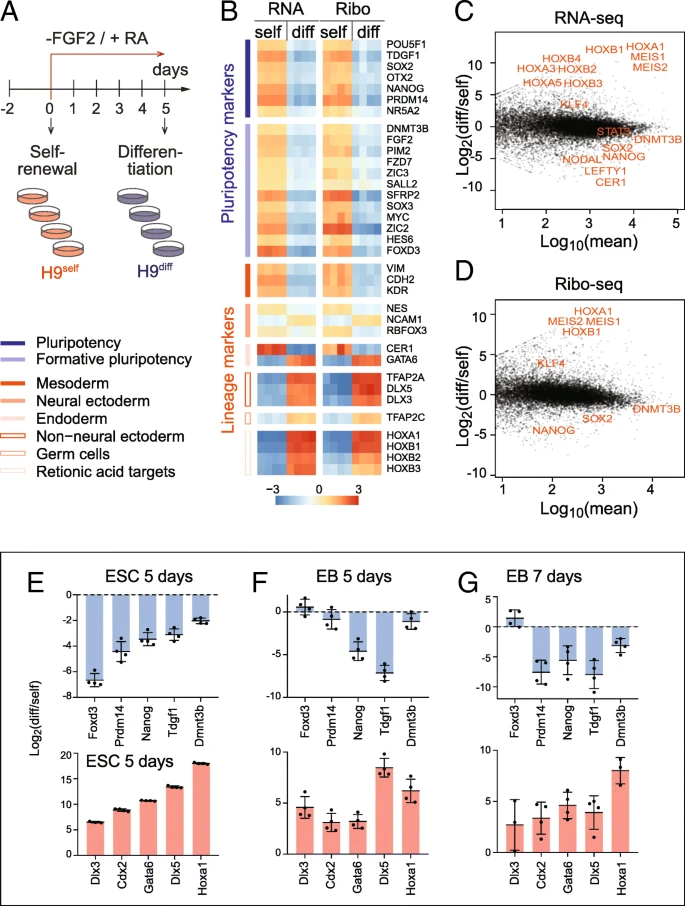
Profiling mRNA expression and translation in self-renewing and differentiating hESCs. a Treatment regime to differentiate the hESCs (H9) by removing FGF-2 from and adding retinoic acid (RA) to the culture medium. Four replicates of self-renewing (H9self) and differentiating (H9diff) hESCs were used for RNA-seq and Ribo-seq analyses. b Expression heatmap of pluripotency (blue) and lineage (red) markers measured by RNA-seq and Ribo-seq. Expression is shown as log2 difference to the mean. Markers in each group are ordered by decreasing expression. c, d MA plots showing the log2 fold change of differentiated (diff) versus self-renewing (self) hESCs against mean expression in either RNA-seq (c) or Ribo-seq (d). Examples of significant genes related to differentiation are shown in red. e–g RT-qPCR confirming similar change in RNA levels of pluripotency markers (upper panels) and lineage markers (lower panels) in hESCs RA-differentiated for 5 days (e) and in embryoid bodies grown for 5 days (f) or 7 days (g). Shown is the mean (n = 3–4). Error bars s.d.
诱导分化5天后,一批维持多能性的marker基因显著下调,而谱系marker显著上调。
After sequencing, we determined the quality of the Ribo-seq data. The library composition of self-renewing and differentiating hESCs was comparable and contained a considerable number of reads mapping to ribosomal RNAs (rRNA), transfer RNAs (tRNAs), and adapter dimers, as expected (Additional file : Figure S1A). We found a large number of genes differentially expressed (Additional file : Figure S1B; Additional file : Table S1). The biological replicates were highly correlated (r = 0.98–1.00) and clustered together (Additional file : Figure S1C). In contrast, the correlation between self-renewing and differentiating samples were much lower (r = 0.70–0.77 in Ribo-seq; r = 0.85–0.88 in RNA-seq). Both the read length distribution and the strong reading frame periodicity for reads of 27 to 29 nucleotides in length confirmed sufficient coverage of mRNAs to analyse translation at single codon resolution (Additional file : Figure S1D-F) []. Metagene analysis further confirmed a strong enrichment of reads over the coding sequences, with only a small number of reads mapping to the 5′ UTRs and nearly no reads at the 3′ UTRs (Additional file : Figure S1G). Finally, the differentially abundant genes in the Ribo-seq and RNA-seq datasets highly correlated (Additional file : Figure S1H), suggesting that transcriptional mechanisms might be the dominant factor changing mRNA levels upon exit from pluripotency [].
Induction of stem cell differentiation using RA in the absence of FGF-2 induced transcripts of all primary germ layers (Additional file : Table S2). Both RNA-seq and Ribo-seq confirmed the existence of heterogeneous cell populations expressing markers of ectoderm, endoderm, and mesoderm, while pluripotency markers were consistently repressed (Fig. b-d) [,,,]. The upregulated genes further included the Hox family, which is known to be regulated through RA-signalling in early embryonic development []. To further confirm that we efficiently differentiated the hESCs, we also grew hESCs in suspension to induce their differentiation into embryoid bodies (EBs) for 5 and 7 days []. The change of mRNA levels of pluripotency and lineage markers were comparable to RA-induced differentiation (Fig. e–g). Thus, RA-treated hESCs exited the pluripotent state and underwent cell differentiation.
Codon composition of cell state-specific mRNAs is biased towards GC content 细胞状态特异性mRNA的密码子组成具有GC含量偏好性
We next asked whether self-renewing and differentiating cells optimized their translational programmes by using cell state-specific codons 自我更新及分化的细胞是否偏好于采用各自细胞状态特异性的密码子?. First, we selected all well-annotated coding sequences from the consensus coding sequence project []. Then, we calculated the relative codon frequency of each gene 每个基因中的相对密码子频率; thereby, each gene was represented as vector of 64 codon frequencies. Using our data, we defined two groups of genes: (i) significantly upregulated genes in self-renewing hESCs and (ii) significantly upregulated genes in differentiating hESCs, and then calculated the overall codon usage compared to all genes (Fig. ).
选择出1.在自我更新的ESC中显著上调的基因,2.在分化过程中显著上调的基因。计算他们的密码子使用频率跟所有密码子进行比较
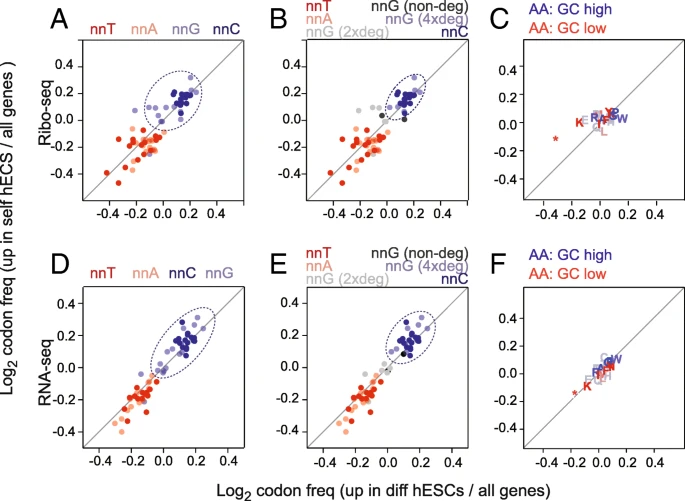
Genomic GC content influences codon usage. a–f Overview of codon (a, b, d, e) and amino acid (c, f) enrichment in differentially expressed genes measured by Ribo-seq (a–c) and RNA-seq (d–f). Enrichment was calculated as log2 fold change of codon or amino acid frequency in differentiation or self-renewal genes relative to all genes. Codons are colour coded according to their third nucleotide (a, d) and are further separated by nnG codon degeneracy (b, e). Dotted circles highlight the most enriched codons in response to the retinoic stimulus
We reasoned that if codon bias contributed to altering mRNA translation between self-renewing and differentiating cells, then it would be most likely to occur at the wobble positions due to the relaxed base pairing and the degeneracy of the genetic code. Therefore, we subdivided the codons according to the base at the wobble positions (nnA, nnT, nnC, and nnG) (Fig. a). The codon enrichment ratios were highly similar for both the upregulated genes in self-renewing and differentiating hESCs (Fig. a). Genes that changed most in expression upon retinoic acid treatment were commonly enriched in G and C at the wobble position (Fig. a; dotted circle) and depleted in A and T. While nnC codons clustered closely together, nnG codon were more diffuse; this was correlated to the codon degeneracy. Only four-fold degenerate nnG codons clustered closely with nnC codons 只有四倍简并的nnG密码子与nnC密码子紧密聚集 (Fig. b). Next, we asked whether the codon bias correlated with a different amino acid usage in the two cell states. However, neither up- nor downregulated genes showed an enrichment for a specific amino acid usage (Fig. c).
The use of optimal codons can enhance mRNA stability and translation, and this effect is evolutionary conserved [,,,]. To address whether optimal codons enhanced mRNA stability in the two cell states, we calculated the relative enrichment of stabilizing and destabilizing codons (Additional file : Figure S2A) using a codon classification made in zebrafish []. The upregulated mRNAs in both conditions showed a slight depletion of strongly destabilizing codons (Additional file : Figure S2B). When we measured the difference between the self-renewing and differentiating genes, we found that upregulated mRNAs in self-renewing hESCs contained significantly fewer destabilizing codons and slightly more highly stabilizing codons (Additional file : Figure S2B; right-hand panels). The association of GC content at the third nucleotide of a codon to high levels of mRNAs was however more pronounced than the association to stabilizing and destabilizing codons 然而,与稳定和去稳定密码子的关联相比,密码子第三核苷酸的GC含量与高水平mRNA的关联更为明显 (Additional file : Figure S2C, D).
GC content defines groups of genes with common function GC含量定义了具有相似功能的基因集
Underlying GC content can substantially contribute to codon usage differences among different cell states [, , ]. We therefore analysed codon usage in genes belonging to various functional categories, including those deployed during self-renewal and differentiation of hESCs. We selected all well-annotated coding sequences and grouped them according to their Gene Ontology (GO) terms. We calculated the average of codon frequencies per GO term and used principal component analysis (PCA) to reveal the underlying sources of variance. The GO terms were primarily separated by GC content (Fig. a); indeed, the first principal component closely corresponded to the GC content of the third codon base (Fig. b). The second principal component reflected the GC content at the first and second bases (Fig. c). Accordingly, genes belonging to similar GO terms such as cell differentiation and neuron fate commitment had similar and high GC content (blue) (Fig. c; right panels).
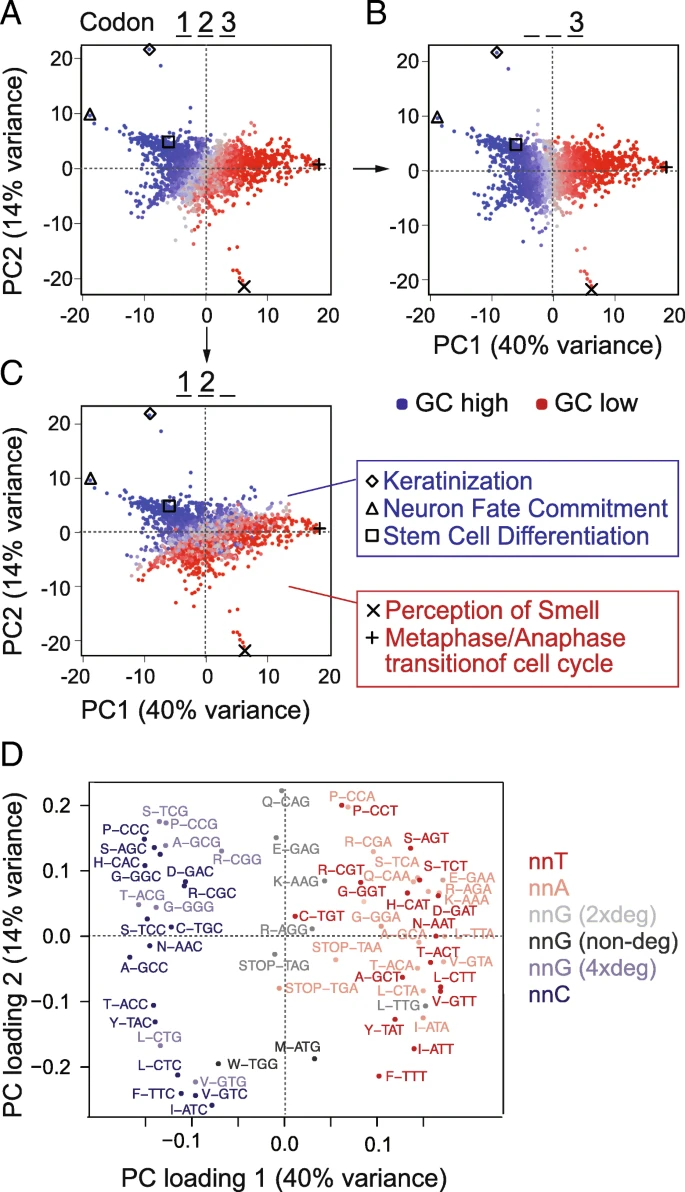
Genes from different GO categories show different codon frequencies. a–c Codon frequencies were calculated per Gene Ontology (GO) term, and PCA was used to reveal a global separation into high and low GC genes (a). GO terms are coloured according to total GC (a), GC at the third codon position (b), or at the first and second position (c). Examples of GO terms are listed next to the graphs. d PCA loadings plot identifying the codons contributing the location of the GOs in a–c. PCA载荷图可识别在a–c 中贡献GO位置的密码子 Codons are colour coded by third position base and/or codon degeneracy. The separation across the x-axis is comparable to the separation across the diagonal in Fig. a, b, d, e
根据每一个GOterm中的基因计算密码子频率,进行PCA。(a)根据每个GOterm中全部密码子的GC含量进行着色。(b)根据第三位密码子的GC含量着色。(c)根据第1,2位密码子的GC含量着色
(d)在PCA图中展示每个GOterm上贡献最大的密码子。
It was unexpected that all three codon positions separated the GO terms according to GC content. While the third and variable base position of codons can be readily varied to optimize translation rates without impacting the encoded amino acids [], changes in the first and second positions almost always result in a different amino acid. Nevertheless, we found that genes belonging to different functional categories were characterized by different codon usage and that this correlated with GC content.
Using a PCA loading plot, we identified the codons driving the global separation by GC content (Fig. d). Codons with cytosine at the third position contributed to the left and codons with adenosine or thymidine at the third position contributed to the right of the plot. As described for our own differentiation data (Fig. b, e), only four-fold degenerate codons with guanosine at the third position contributed to the left of the PCA plots (Fig. d). The high level of similarity between the global PCA analysis of all genes and our differentiation experiment suggested that genes with similar functions share similar inherent genomic features. In conclusion, genomic GC content influences codon choice in the differentiation gene expression programme.
Self-renewing ESCs share a common codon signature at the ribosome A- and P-sites 自我更新ESC在核糖体A位点和P位点共享类似的密码子信号
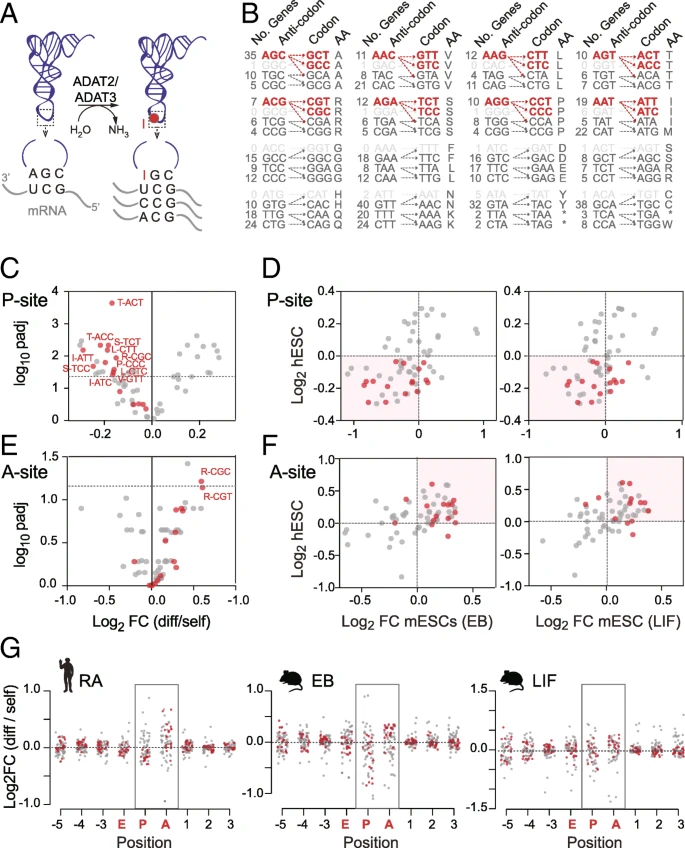
While GC content was one factor for codon usage optimization, protein translation efficiency is likely to be regulated by additional factors. The translation kinetics of codon-anticodon pairs in the ribosome are complex and determined by the availability and binding affinity of matching and mismatching tRNAs [, , ]. In part, the translation rate is directed by the competition between near-cognate and cognate aminoacyl-tRNAs entering the ribosome 翻译速率由进入核糖体的近同源和同源氨基酰基-tRNA之间的竞争指导. Therefore, we asked whether self-renewing and differentiating hESCs showed differential codon stalling at the active sites of the ribosome.
During translation, only three codons actively pair with tRNAs, known as the ribosome E-, P-, and A-sites (Fig. a). Cognate aminoacyl-tRNAs enter the ribosome at the A-site. After transfer of the polypeptide chain to the tRNA, the tRNA moves from the P- to the E-site and is then released from the ribosome. Although the ribosome only actively engages with three codons within an RNA, it covers more than 27 nucleotides (Fig. a). Due to the three-nucleotide periodicity reflecting the triplet nature of the genetic code (Additional file : Figure S1E, F) [], the actively bound codons at the E-, P-, and A-sites can be calculated by counting nucleotide triplets from the 3′ end of the read fragments. 在E-,P-,A-位点的活跃结合密码子可以通过计算read3‘端的三联核苷酸获得 We numbered the ribosome-protected codons from − 5 to + 3, with 0 corresponding to the A-site (Fig. a). 我们给核糖体保护的密码子从-5到+3进行了编号,0代表A-位点 We first calculated the codon frequencies per footprint position 我们首先计算了每个印记位置的密码子频率. To do so, we compared the relative frequency of a codon at the ribosome P- (or A-) site to the frequency of this codon in all Ribo-seq reads from the sample. 我们的做法是,通过在P-位点的密码子的相对频率,比上这一密码子在Ribo-seq样本中所有reads的频率
11
Distinct codon signature at the ribosome A- and P-sites in self-renewing ESCs. a Illustration of a ribosome binding to mRNA protecting at least nine codons (position − 5 to + 3) from degradation by the RNase digestion. Positions − 2, − 1, and 0 correspond to the E-, P-, and A-site that host the charged tRNA. b The raw codon frequency was calculated for each specific codon (e.g. A-site) and normalized to the mean frequency across all nine positions. 计算了每一个特定位点(A-位点)的初始密码子频率,并且根据所有9个位置进行了归一化 Calculations were done separately for self-renewing and differentiating samples. c–e Codon enrichment at the E- (c), P- (d), and A-sites (e). Stop codons are highlighted in red, and proline codons are highlighted in blue. The codons are sorted according to their frequency (shown in Additional file : Figure S3A-C). f, g Codon enrichment of stop (f) or all other codons (g) across all ribosome-protected codons. h Log2 fold change of normalized codon usage in differentiated versus self-renewing hESCs across all ribosome-protected codons. Significantly different codons (Welch’s t-test; FDR correction) are marked in red (enriched) and blue (reduced) and occur mainly at the P-site. i Log2 fold change of normalized codon usage in mouse differentiated (embryoid bodies) versus self-renewing embryonic stem cells (mESCs). Blue and red dots represent the significantly changed codon shown in h
As expected, some codons occurred more frequently than others (Additional file : Figure S3A-C). Since the codon frequencies at the positions flanking the tRNA-bound sites (− 5 to − 3, and + 1 to + 3) are expected to follow the genomic codon distribution, they served as a negative control. We then normalized the raw frequencies to the frequency across all nine positions (Fig. b). This will correct for differences in mRNA abundance, and neither transcriptional nor differences in mRNA decay should affect the following analysis. The strong enrichment of stop codons at the A-site and their depletion at the P- and E-sites validated this approach (Fig. c–e). Although Proline incorporates more slowly in translation [], its codons were not strongly enriched at the A-, P-, or E-site 尽管脯氨酸在翻译中的融合速度较慢[46],但其密码子在A,P或E位上并未强烈富集 (Fig. c–e). When we compared the enrichment of codon frequencies along all sites in the ribosome, we found the highest variation at the A-site (Fig. f, g), indicating that the stalling at the A-site was rate limiting for translation elongation 当我们比较沿核糖体所有位点的密码子频率富集时,我们发现在A位点的变异最大(图4f,g),这表明在A位点的停滞是翻译延伸的速率限制 [, ].
Next, we tested whether the normalized codon frequencies differed in self-renewing and differentiating hESCs (Fig. h). We observed differences at both the A- and the P-sites, yet most significant differences occurred at the P-site (Fig. h; Additional file : Figure S3D). To test whether the codon usage differences in human ESCs was an evolutionarily conserved feature of translational control, we performed similar analyses using published Ribo-seq and RNA-seq data obtained from self-renewing and differentiating mouse embryonic stem cells (mESCs) []. The exit of pluripotency and differentiation of the mESCs was induced using two well-established methods: the withdrawal of leukemia inhibitory factor (LIF) from the culture medium and by growing the mESCs as embryoid bodies (EBs) [,,]. As described for hESCs, we excluded rRNA and tRNA reads and selected read lengths with high reading frame periodicity (Additional file : Figure S4A-E). Again, most differences occurred at the A- and P-sites (Fig. i). Although the mouse data were noisier, the significantly depleted codons at the P-site in differentiated hESCs were often also less common in differentiated mESCs (Fig. h, i; Additional file : Figure S4F; blue dots). Thus, we found a distinct set of codons with a higher likelihood of being stalled at the ribosome P-site in mouse and human self-renewing embryonic stem cells. 因此,我们发现了一组独特的密码子,它们更有可能停滞在小鼠和人类自我更新胚胎干细胞的核糖体P位点。
A prominent self-renewing codon signature is hetADAT-sensitive
A key factor modulating codon translation is tRNA selection []. To enhance codon binding, the tRNA wobble nucleoside (position 34) frequently carries chemical modifications []. One such prominent and essential tRNA modification is wobble inosine [, ]. Inosine is formed by a deamination reaction of adenosine and catalyzed by the heterodimeric enzyme adenosine deaminase acting on tRNA (hetADAT) composed of two subunits ADAT2 and ADAT3 (Fig. a) []. While tRNAs carrying adenosine at position 34 (A34) pair with uracil, inosine (I34) is capable of pairing with adenosine, uracil, and cytosine and thereby enhances translation efficiency (Fig. a) [, ]. In eukaryotes, the modification is present in eight different tRNAs that recognize the eight nnT codons. Inosine in tRNAs is required to translate nnC codons that have none or few tRNA gene copies (Fig. b) []. In principle, the same tRNAs can also bind eight nnA codons, yet these codons are also translated by their own complementary tRNAs.
Distinct codon usage signature in pluripotent cells are hetADAT-sensitive codons. a Illustration of hetADAT (ADAT2 and ADAT3)-mediated adenosine-to-inosine tRNA editing at position 34 in the anticodon loop and the codons read by unmodified and modified tRNAs. b HetAD; self-renewing hESCs. HetADAT-dependent codons are highlighted in red. Correlation (d, f) between log2 fold change of codon usage frequencies in differentiated human (hESCs) with differentiated mouse (mESCs) embryonic stem cells through embryoid body culture (EB; left-hand panel) or by removal of LIF (right-hand panel) at the P-site (d) and A-site (f). g Log2 fold change of codon usage frequency across all sites protected by the ribosome in human (left-hand panel) and mouse ESCs differentiated through EB culture (middle panel) or removal of LIF (right-hand panel). HetADAT-dependent codons are highlighted in red
We noticed that codons showing significant differences in self-renewing hESCs were often translated by inosine-modified tRNAs (Fig. c). All nnT and nnC codons that depended on inosine tRNA for translation occurred less often at the P-site in differentiated hESCs (Fig. c). Moreover, these differential codon frequencies highly correlated between human and mouse (Fig. c). Thus, mouse and human differentiating ESCs showed a depletion of hetADAT-dependent codons at the P-site. 因此,小鼠和人的分化ESC在P位点显示出依赖于hetADAT的密码子的耗尽。 As described above, due to the high variability of codon frequencies at the A-site, we were unable to identify significant changes (Fig. h). However, when we now plotted the A34I-dependent codons, we found the vast majority of them enriched at the A-site in both mouse and human differentiating ESCs (Fig. e, f; Additional file : Table S3a,b). Finally, we confirmed that this distribution was specific to the A- and P-sites and did not occur at any other site in the ribosome (Fig. g). In summary, we found that in self-renewing ESCs hetADAT-dependent codons occurred less often at the ribosome A-site but more often at the P-site when compared to differentiated ESCs.
The time it takes the ribosome to ensure that a tRNA in its A-site is indeed a cognate tRNA for the codon, is considered rate limiting for translation elongation [, ]. In addition, whenever a ribosome is bound to an mRNA waiting for a cognate tRNA at its A-site, a tRNA is already bound to its P-site, attached to the growing polypeptide chain. Thus, the lower frequency of hetADAT-dependent codons at the A-site indicated their faster translocation to the P-site in self-renewing ESCs.
hetADAT regulates codon usage in self-renewing embryonic stem cells
The lower frequencies of hetADAT-dependent codons at the A-site of ribosomes in self-renewing ESCs might indicate an enhanced activity of the catalytic complex. Indeed, the mRNA level of ADAT2 was significantly higher in self-renewing hESCs when compared to RA-differentiated cells or EBs (Fig. a). To quantify the levels of inosine in hetADAT-dependent tRNAs, we performed tRNA sequencing in self-renewing and differentiated hESCs (Additional file : Table S4). We used the fact that inosine will form the strongest bond to cytosine and is therefore reported as a guanine during sequencing. The level of A34I modifications can thus be quantified by measuring the A-to-G substitutions at the anticodon wobble position (Additional file : Table S5). The A34I tRNA modification levels were significantly higher in self-renewing (83–86%) than differentiated (60–79%) hESCs (p = 0.031; Student’s t test) (Fig. b). Accordingly, the A34I modification occurred less often in the majority of hetADAT-dependent tRNA isotypes (Fig. c). Thus, self-renewing hESCs have higher levels of A34I tRNA modifications than differentiating cells. 因此,自我更新的hESC比分化细胞具有更高水平的A34I tRNA修饰。
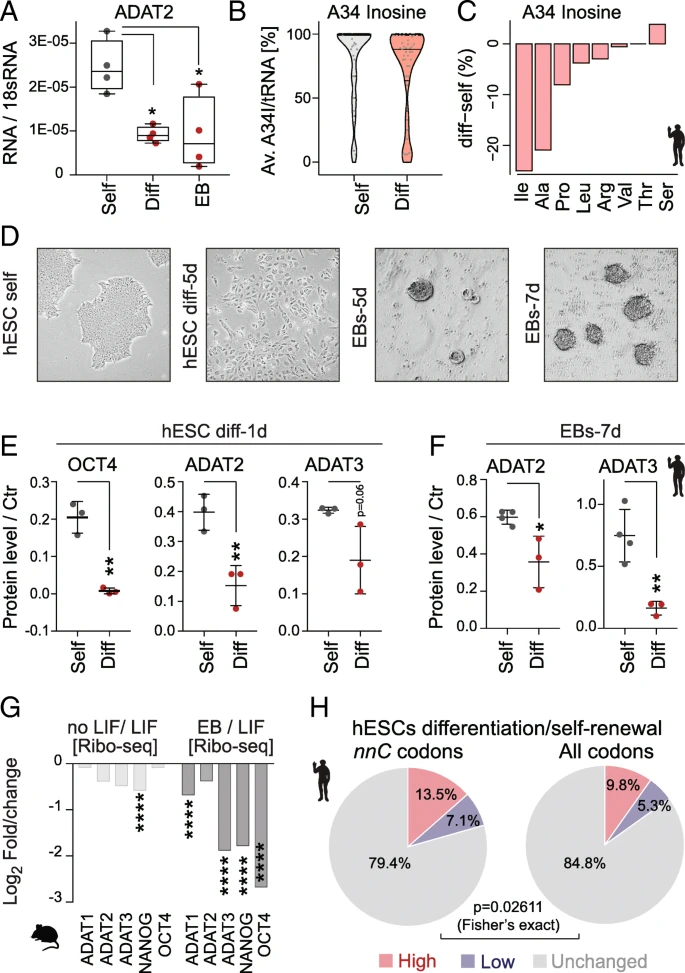
HetADAT-dependent translation in mouse and human ESCs. a RT-qPCR confirming downregulation of ADAT2 mRNA levels in differentiated hESCs (Diff) and embryoid bodies (EB) compared to self-renewing hESCs (Self). * p < 0.05 one-way ANOVA. b Distribution of wobble inosine (A34I) levels per tRNA (> 10 reads) shown as Violin plot with median (black line) and quartiles (dotted line). c Differences of inosine levels at A34 in differentiated versus self-renewing hESC per tRNA isotype. d Light microscopic image of self-renewing (self) human embryonic stem cells (hESCs), differentiated hESCs for 5 days (5d), and embryoid bodies (EBs) after 5 and 7 days in suspension. e Quantification of protein expression using Western blotting of OCT4, ADAT2, and ADAT3 in self-renewing (grey) and differentiating (red) hESCs for 1 day. p < 0.01 unpaired t-test. f* Quantification of protein levels using Western blotting of ADAT2 and ADAT3 after differentiating hESCs into EBs for 7 days (7d). **p < 0.05; p < 0.01 unpaired t-test. g* Log2 fold change gene translation (Ribo-seq) of indicated proteins in mouse self-renewing ESCs versus ESCs differentiated by removal LIF or grown as embryoid bodies (EB). *p < 0.0001. h Ribo-seq fold changes (diff/self) was compared with RNA-seq fold changes (diff/self). Genes with ribosome occupancy difference two-fold higher (red), two-fold lower (blue) or similar (grey) to the mRNA level difference was identified among genes rich in hetADAT-dependent nnC codons (left) and among all genes (right). The hetADAT-dependent group was significantly enriched for genes with altered (higher and lower) ribosome occupancy
Next, we confirmed that also the protein levels of ADAT2 and ADAT3 decreased with differentiation of hESCs (Fig. d–f; Additional file : Figure S5A-E). ADAT2/3 protein expression was significantly reduced after only 1 day of treatment with retinoic acid (Fig. e; Additional file : Figure S5D). Downregulation of hetADAT was further confirmed when the hESCs were grown in suspension to induce their differentiation into EBs (Fig. f; Additional file : Figure S5E). Similarly, translation of mouse ADAT2 and ADAT3 was downregulated upon differentiation, yet the differences were only significant when compared to differentiated EBs (Fig. g).
Finally, we asked whether the optimized codon usage in self-renewing ESCs resulted in differential translation of hetADAT-dependent codons. 是否在自我更新的ESC中采用了优化后的密码子导致了hetADAT依赖的密码子翻译的差异 We calculated the total frequency of the eight nnC codons that rely on the inosine modification for translation in all genes 我们在所有基因中计算了全部8个依赖于肌苷修饰的nnC密码子频率 (Fig. b). Then, we selected all coding sequences containing more than 28.4% (top 1%) nnC codons and asked whether they showed translational differences in self-renewing versus differentiated hESCs that were independent of mRNA levels 接下来,我们选择所有包含nnC密码子超过28.4%的编码序列,并且询问他们是否在自我更新和分化的hESC中显示出不依赖mRNA水平的翻译差异 . Comparing the two cell states, we found that translation of 13.5% nnC-high coding sequences was enhanced and 7.1% repressed (> 2-fold) (Fig. h; left panel). This difference in translation of nnC-enriched genes was significantly different when compared to all coding sequences (Fig. h; right panel). Thus, the differential translation of nnC-high coding sequences in self-renewing hESCs cannot be explained by differences in RNA abundance alone.
Together, our data suggest that self-renewing ESCs translate hetADAT-dependent codons with higher efficiency when compared to differentiating ESCs. This difference in hetADAT-dependent codon usage occurred independently of the nature of the differentiation stimulus and was conserved in mice.
Discussion
Protein synthesis is a fundamental process in all cells, yet recent studies revealed distinct regulatory functions of the mRNA translation machinery in stem cells. Low global protein translation rates are commonly found in adult undifferentiated stem and progenitor cells and are required to maintain a fully functioning stem cell state [,,]. Mouse embryonic stem cell differentiation also correlates with an overall increase in protein synthesis and enhanced mRNA loading into polyribosomes []. One explanation for the differences in protein synthesis might be the expression of distinct tRNA pools that optimize codon usage in different cell states []. However, the use of tRNA concentrations to understand codon usage is hampered by the fact that in addition of being crucial adaptors during translation, tRNAs are important regulators of a wide range of biological processes []. An alternative method to analyse codon usage is ribosome profiling, which has been applied to various organisms and cell contexts, for example amino acid starvation, oxidative stress, the perturbation of signalling pathways, and embryonic stem cell differentiation [, ,,,].
Here, we use ribosome profiling to analyse the importance of optimized codon usage in regulating gene expression in different cell states. Our work identifies a codon bias in self-renewing and differentiating stem cells, and we propose that this is driven by both mRNA levels and translational mechanisms. Genes belonging to the same functional category use similar codons characterized by differences in GC content. Since we measured similar GC content enrichment in up- and downregulated genes, our observed differences are unlikely to be directly linked to mRNA degradation. Our data is consistent with those of previous studies in Neurospora, where codon usage strongly correlated with protein and mRNA levels genome-wide but was largely independent of mRNA translation and stability [].
Thus, GC content is one important factor that influences codon choice during gene expression. The concept that codon optimization is due to the GC content of the underlying genes has been demonstrated before for both specific genes and genome-wide [, , ]. However, we further propose that the regulation of the protein synthesis machinery through chemically modified tRNAs plays important additional roles in regulating gene expression programmes. We find that codons whose translation depends on hetADAT-modified tRNAs were under-represented at the ribosome A-sites in self-renewing mouse and human ESCs. We provide further evidence that a faster translation of hetADAT codons in self-renewing cells might be facilitated by higher levels of inosines at the tRNA wobble position. While a role for hetADAT in embryonic stem cell fate has not been described before, it is well known that tRNA wobble modifications ensure efficient protein synthesis, maintain protein homeostasis, and promote cell adaptation and survival [].
The existence of a nuclear (GC content) and a cytoplasmic (translation) mechanism that both determine codon usage during stem cell differentiation raises the question of how they are connected. It is plausible that a cytoplasmic tRNA modification-driven codon bias may help to overcome a GC-content-driven codon usage for cell type-specific mRNAs, thereby enhancing translation of certain groups of mRNAs. However, the functional analyses on how precisely hetADAT regulates codon usage and how this influences gene expression in pluripotent embryonic stem cells is hampered by the fact that ADAT2/3 is essential for cell survival []. Knock-down of hetADAT causes cell death in human cells. Over-expression also affects cell survival because ADAT2/3 is a highly mutagenic enzyme []. Therefore, ADAT2/3-mediated C to U deamination at position 32 requires the prior formation of 3-methylcytosine in Trypanosoma brucei []. Thus, increasing the hetADAT levels might not be sufficient to increase inosines specifically at the wobble positions.
Together, we provide evidence for an hetADAT-dependent codon bias in self-renewing embryonic stem cells that might suppress differentiation and lineage commitment.
Conclusion
In this study, we used RNA-seq and Ribo-seq to decipher transcriptional and translational mechanisms regulating codon bias in self-renewing and differentiating human embryonic stem cells. We revealed that codon usage during stem cell differentiation is regulated at the mRNA levels and during translation. We confirm that codon usage of differentially expressed genes is primarily characterized by genomic GC content.
https://blog.sciencenet.cn/blog-565558-1260533.html
上一篇:2020-11-24=codon-usage
下一篇:2020-12-12=Condon Usage