博文
介绍QVASP在表面吸附计算中的一个小技巧
||
计算表面吸附分子体系的时候,我们会构建一个表面,然后把分子放在这个表面上的不同位点上,构建出一系列的结构,然后提交到VASP里面去计算,最后我们需要找到最低能量结构的吸附结构,然后再做进一步的分析使用。
QVASP提供了一个快速的在若干个文件夹里,读取每个结构的能量的方法,处理这类问题非常方便。
比如我计算一个MoN(200)面上吸附一个H2分子,根据不同的吸附位点,有很多不同的吸附构型,把他们都计算了出来。产生了一系列的文件夹。

这时候我只要使用qvasp -e命令,就可以得到如下信息:
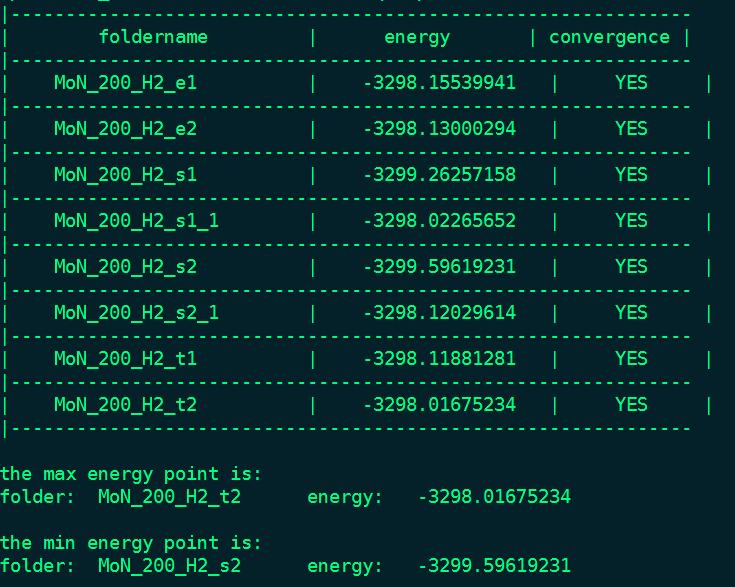
就可以把所有的文件夹里的OUTCAR读取出来,把能量和是否收敛也print 出来,最后,把最低和最高的能量文件夹给出来。这样我们就可以非常方便的提取到我们需要的信息,还可以检查是不是收敛了,因为文件夹一多,有时候自己也会忘记检查。真的非常方便。如果前面的列表能够按照能量大小顺序进行一个排序就更好了。
另外,一个小功能是:qvasp -zpe,可以查看zero point energy(零点能)

运行命令以后,可以得到零点能,而且可以检查下,计算结果里是不是有虚频,如果有虚频也会print出来。
附一下qvasp的网址:https://sourceforge.net/projects/qvasp/
https://blog.sciencenet.cn/blog-478347-1215490.html
上一篇:Latex编译器推荐(面向初学者或者懒得折腾的朋友,主要针对windows用户)
下一篇:推荐一个dos仿真器(ConEmu)