博文
应用 PhyloMad对进化模型准确性进行评估(一)
|||
(一)应用PhyloMad进行核苷酸替换模型准确性的检验
絮语:
系统发育分析方法的准确性通常依赖于选择合适的进化模型,目前很多模型选择软件(jModelest、MrModelTest、ProtTest等)通过对许多候选模型的比较分析进行确定,但评估这些选择的模型是否真是适用于分析的数据集,这些软件无力解决这个问题。
近日有网友HPV的返修文章,审稿人提及该DNA病毒选用的HKY模型可能不适合。
审稿人的意见如下:
Most issues have been addressed. I would still prefer that the methods section for the phylogenetic analysis was expanded. For example, I disagree with HKY being a relevant model for HPV evolution. So please justify this choice.
该HPV数据的最佳模型:
Akaike Information Criterion: TIM+I
Corrected Akaike Information Criterion: TIM+I
Bayesian Information Criterion: HKY
Best-fit model: HKY chosen according to BIC
如何回复这个问题(红色突出显示处),应用PhyloMad对HPV数据进行核苷酸替换模型准确性的检验,或许可以为回复意见提供一个参考(详见文后的参考回复意见)。
本文以HPV数据为例,简明图解一下MEEP课题组博士后 Duchêne 开发的软件-PhyloMad的使用,以飨初学者。特别感谢@西梦飘飘提供示例数据。
所需工具:
PhyloMad: https://github.com/duchene/phylomad
操作图解:
1. 安装PhyloMad
根据软件官方的提示,下载后释放一个本地目录,同时需要安装一些程序运行所需要的R包,包括shiny、methods、phangorn、ape、foreach、doParallel、iterators、parallel、phytools、maps和apTreeshape等。只需双击目录内的 phylomad.Rscript,它会自动下载相关的包,此处不再赘述。
2. 运行PhyloMad
Mac 系统下,双击 runMac.command即可启动 PhyloMad;而Windows系统下,双击runWin.vbs即可运行。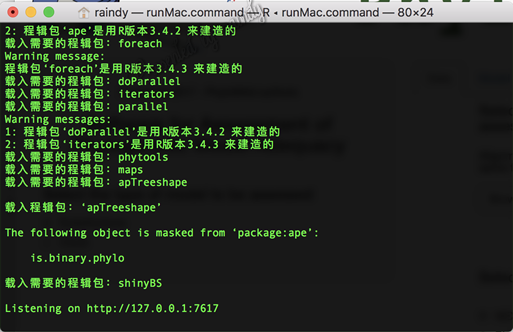
程序自动弹出终端或命令行界面,同时弹出一个网页界面,参数设置主要在网页界面中设置完成,而实时运行的状态则在终端或命令行中显示。
3. 载入序列
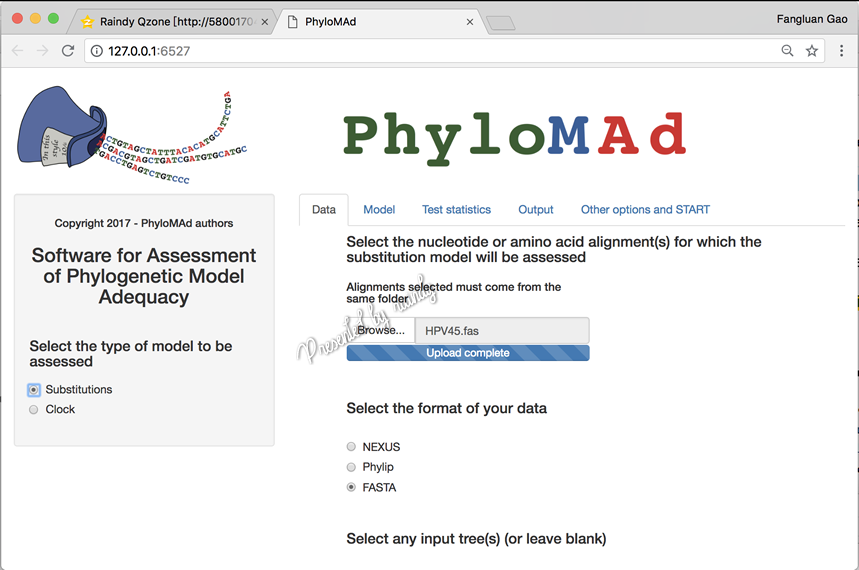
PhyloMad支持两种类型的模型准确性评估,一种位点替换模型,另一种是分子钟模型(当前只支持BEAST2.x系列,不支持BEAST1.x),本实例是核苷酸替换模型的准确性评估。因此,界面左下角的模型类型选择为“Substitutions”
在网页界面中的“Data”标签下,通过“Browse...”载入序列,并选择该序列的正确格式(此处若选择不对,容易报错无法运行)。Tree文件可以软件自己生成,也可以将自己建树的树上传,同样选择该文件正确的格式。
4. 评估模型
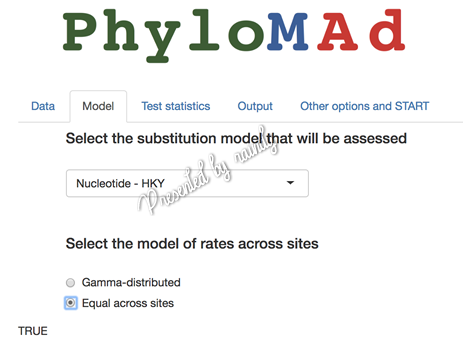
切换到“Model”选择要评估的模型,本实例数据由ModelFinder获得的最佳模型为HKY,故而此处选择此模型进行检验,在下拉菜单中选择“Nucleotide - HKY”,如果位点间的速率是呈Gamma分布 (即:+G),复选框为Gamma-ditributed;如果位点间的速率都一样,则选择“Equal across sites”,即本实例数据的情形。
5. 统计检验
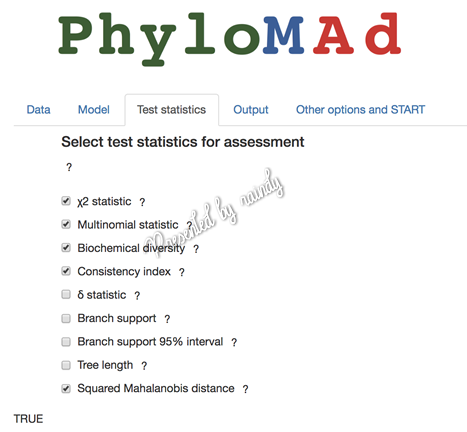
核苷酸数据的模型检验主要包括常见的五种,分别卡方、多项分布、生化多样性、一致性指数和卡方马氏检验等,此五项检验为默认选中状态。
6. 结果输出
每个位点的准确性指数(Metrics of adequacy for individual loci)和检验结果绘图(Test plots)为默认项目,可以不用修改。

7. 其他参数
数据模拟数,默认为100,可以不用修改;多核CPU的计算机选择用于分析的内核数默认值为1,用户根据实际调整。
配置完毕,点击“Start Assesement” 即可开始评估。
100个模拟数据运行完成,终端或命令行界面最下方出现运行用时(Time used ...)说明运行完成。
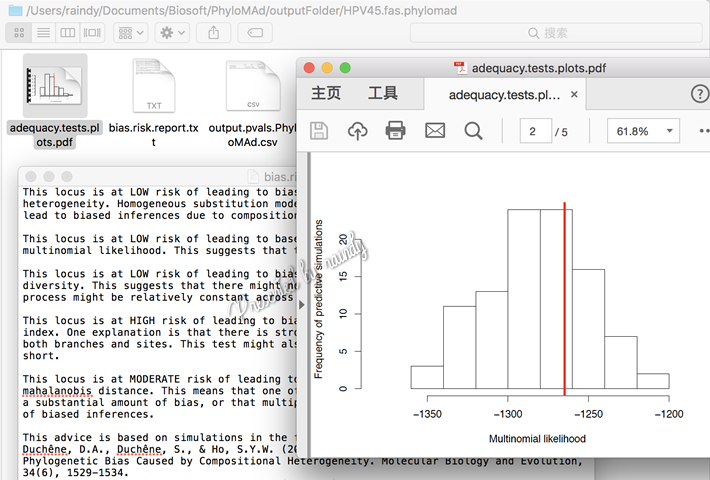
8. 结果解读:
PhyloMad 评估模型准确性完成后,会生成三个文件,一个是五个检验的Plots图,一个是使用该模型的风险评估报告,最后还有一个不同检验的具体值。
参考回复意见:
We share the reviewer’s concerns about model misspecification and have added a test of substitution model adequacy using PhyloMAd. Our test is based on a newly published approach and uses five test statistics to examine the potential impacts of model inadequacy on phylogenetic inference. Our analysis confirms the adequacy of the HKY substitution model used in our analyses.
参考文献:
1. Duchêne, D.A., Duchêne, S., Ho, S.Y.W., 2018. PhyloMAd: Efficient assessment of phylogenomic model adequacy. Bioinformatics, In press.
2. Yang, Z., Zhu, T., 2018. Bayesian selection of misspecified models is overconfident and may cause spurious posterior probabilities for phylogenetic trees. Proceedings of the National Academy of Sciences, In press.
注:本文首发于QQ空间日志,欢迎转载,但请保留作者信息:https://user.qzone.qq.com/58001704/blog/1530005771
https://blog.sciencenet.cn/blog-460481-1121537.html
上一篇:BEAST 分析 FAQ (20180302更新)
下一篇:日期随机化检验(DRTs)之 TipDatingBEAST篇
全部作者的其他最新博文
全部精选博文导读
相关博文
- • AAA PCI -- 学者心声(Authorea 预印论文报道)
- • Building a DIKWP-TRIZ Software System (初学者版)
- • DIKWP-TRIZ: Semantic Blockchain and Semantic Communica(初学者版)
- • DIKWP-TRIZ in 3-No Problem and Artificial Consciousnes(初学者版)
- • Comparison Between DIKWP-TRIZ and TRIZ(初学者版)
- • DIKWP-TRIZ: Enpower AI/AC Innovation (初学者版)