博文
BEAST 分析 FAQ (20180302更新)
|||
Raindy 注:
本文首发于本人QQ空间日志(https://user.qzone.qq.com/58001704/blog/1502418934),将不 定期更新,希望对初学者有帮助,欢迎关注。如需转载请保留原作者信息。
Q1: BEAST 有1.x 和 2.x 两个系列,如何选择?
A1: 可以根据分析不同目的进行选择,两个版本的特点简述如下:
I、BEAST 1的特点:
(1)适用于分析群体或物种级别的数据;
(2)可以同时评估树和节点的时间;
(3)分子钟模型的范围更广;
(4)树先验(Tree Prior) 和 demographic 模型范围更广;
II、BEAST 2的特点:
(1)重写的程序,更多模块化;
(2)可以通过添加包(Packages)来扩展BEAST;
(3)BEAST 1中的树先验(Tree Prior) 在该版本中不可用;
(4)支持数据模拟;
更多的功能比较,可以访问BEAST官网查看(http://beast2.org/beast-features/)
Q2: 如何比较BEAST中哪个模型最佳?
A2 : 一般通过贝叶斯因子法(Bayes Factor, BF)来比较两个模型的边际似然值(Marginal Likelihood),简单办法:通过BF法分析后,哪个模型对应的LnL值越大,相对更优。具体也可以通过以下操作来确定:在Tracer 中同时导入两个模型的xx.log 文件,选中所有要比较的模型后,依次点击菜单Analysis--->Model Comparation,Analysis type改为Harmonic mean,Reps=100,最后根据获得的BF值(BF=Pr(D|M1)/Pr(D|M2)至少大于3,则说明M1>M2)范围判断模型间的优劣程度。下图所示,第三个数据最佳(P_UCL_Constant_80M.log)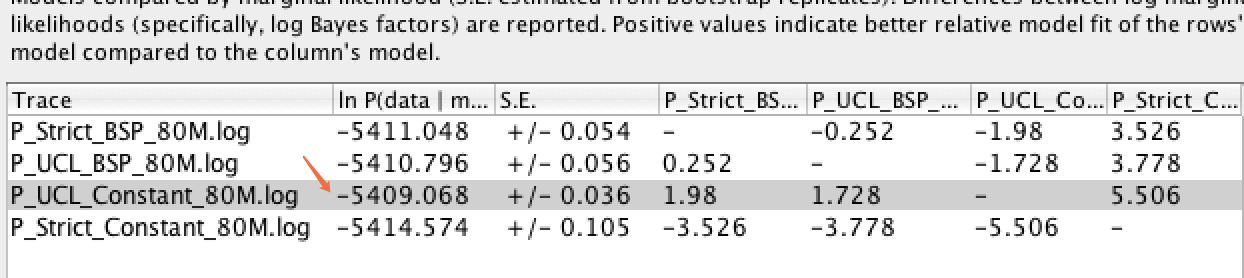
Q3: 模型中位点间的速率变异呈gamma分布,如何确定较为准确的Gamma类型数(Number of Gamma Categoies, nCat)?
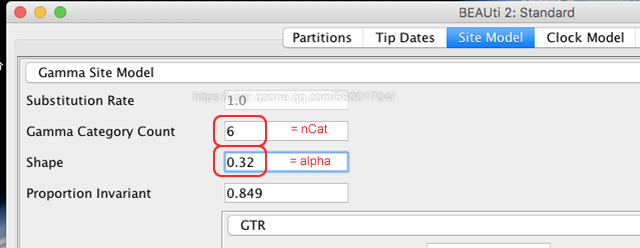
A3: Gamma分布的形状通过alpha参数来确定,如下所示。当alpha值很小时,大部分位点慢速进化,只有一小部分位点快速进化;但当alpha值非常大时,大部分的位点以接近相同的速度进化。因此,当alpha值比较小时(如:shape=0.32,该值可以通过模型软件计算获得),可以将nCat设置为6(默认值为4)进行分析 。
Q4:BEAST 分析过程中,断电了怎么办?
A4: BEAST 1.x 没有断点续行功能,可以更名后重新运行,最后合并前后运行的数据;BEAST 2.x 有断点续行功能,可以追加运行,有点类似于Mrbayes的Checkpoint功能,如下图所示。
Q5:Baysian skyline analysis 时,popsize和groupsize一直不收敛,怎么办?
A5: 这两个参数不收敛,说明BEAUti配置时这两个所使用的默认参数不合适,可以在Operator标签修改(下图)或直接用记事本类工具修改,一般修改为原来值的10倍,如下图所示,将15.0改为150,6.0修改为60即可...此方式也适用于 Exponential growth 模型。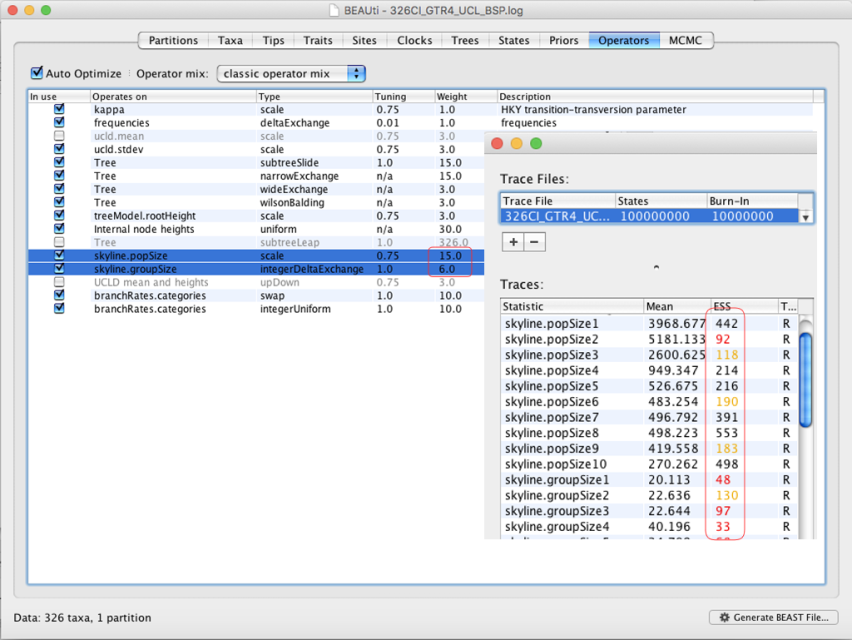
Q6:如何给BEAST及相关的工具增加虚拟内存?
A6: BEAST运算生成的结果文件,特别是tree文件,通常都比较大,使用TreeAnnotator 生成MCMC树,经常因为内存不足报错,其主要原因是虚拟内存设置过低。解决的办法有两种,一种是直接修改Java程序包中的Info.plist文件;另一种是用命令行运行,类似“java -Xms64m -Xmx256m -jar lib/beast.jar”。前者修改后一劳永逸,推荐使用,选择程序对象,如BEAST后,右键选择“显示包内容”,在打开的“Contents”目录内用文本编辑器工具打开Info.plist(mac系统推荐使用免费Pref setter)将JVMOptions下的Item 1的虚拟内存调大,如 -Xmx256m改为-Xmx6g,如下图所示:
Q7:如何判断自己的数据适合严格(strict)分子钟还是宽松(relaxed)分子钟模型?
A7: 可以先用宽松分子钟结合简单的Tree piror配置个xml文件,MCMC链长不需要很大,运行结束后,将xx.log文件通过Tracer导入,查看xxx.coefficientOfVariation 这个参数,该值介于0和1之间。如果值越接近于0,说明数据更接近严格分子钟模型;反之,更偏向于宽松分子钟模型,如下图,该值为0.511,说明该数据适合宽松分子钟模型。
PS:如果分析的数据有时间信号,可以通过Phylogenetic dating的treedater包来分析,详见日志:http://user.qzone.qq.com/58001704/blog/1506493780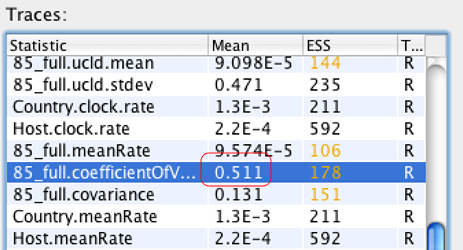
Q8:如何判断数据中的采样时间是否可用于分子钟校准?
A8: 一般启用BEAUTi中的Tip dates 功能前,需要做个日期随机化检验(Date-randomizationtests, DRTs);如果没有通过DRTs,则说明数据中没有时间结构,不可用于分子钟校准。常见的DRTs 分析软件有TipDatingBeast(R包)、least-squares dating (LSD) ,如下图所示,该数据的采样时间具有时间信号,可以用于分子钟校准,相关方法描述详见本人2017年发在EVA上的文章(http://onlinelibrary.wiley.com/doi/10.1111/eva.12459/abstract)。
� �
�
Q9:如何绘制 Extended Bayesian Skyline Plot 图?
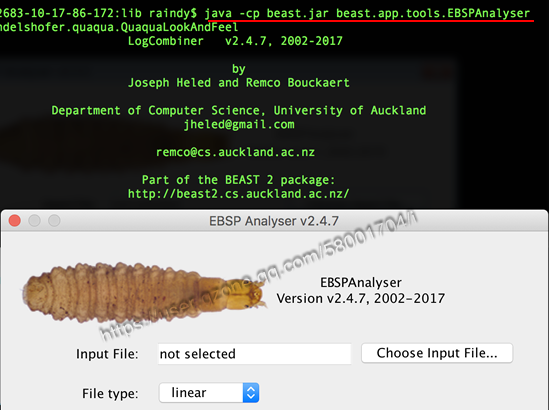
A9: 常规的Bayesian Skyline 可以直接通过Tracer 的Analysis菜单中“Bayesian Skyline Reconstruction”来生成,然而Extended Bayesian Skyline Plot (EBSP) 却不能直接应用Tracer操作,早期绘制EBSP图也比较麻烦,通常需要一些脚本来实现。其实一些高版的BEAST已经内置EBSP analyser,只是需要通过命令行来操作,可以通过cd 命令切换到beast.jar 所在的目录后,输入 java -cp beast.jar beast.app.tools.EBSPAnalyser即可调用出EBSP analyser,如下图所示。如果喜欢用R脚本,当然可以下载 RplotEBS.R(https://sites.google.com/site/santiagosnchezrmirez/home/software/r)来操作
� �
�
Q10:如何获得MCC树中的根状态后验概率(root state posterior probabilities)?
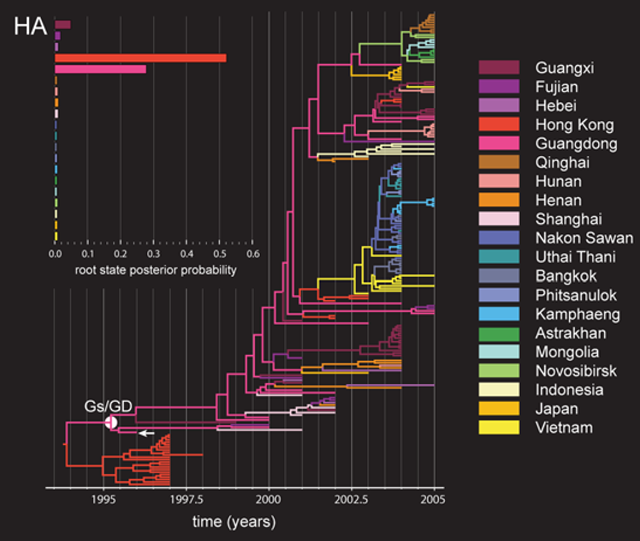
A10:可以打开 TreeAnnotator 对定义Trait的Sample tree进行注释,最后得到MCC树,用Notepad++之类的工具打开,并将光标定位到文件末尾,找到一串类似于“xxx.set.prob”的字符串,=后紧随的是各定义Trait的后验概率数值,对应的Trait 顺序可以向后找到“xxx.set=”,比如:Country.set.prob={9.777E-4,0.989,...}, Country.set={"China","Turkey","Spain",...} 即中国、土耳其、...对应的root state posterior probabilities分别为9.777E-4,0.989,...。这些数据提取到Excel中通过绘制簇形柱形图即可得到类似下图左上角的效果图。
� �
�
Q11:如何通过LSD快速获得tMRCA和进化速率?
A11:估算tMRCA和进化速率常见的方法有:(1)Bayesian算法(软件:BEAST);(2)Least-squares dating(软件LSD);(3)Root-to-tip (软件TempEst);(4)Treedater (R包Treedater);(5) TreeTime (Python 包)。其中BEAST最为常用,但其运算量相对较大。相比较之下,LSD以快速著称,可以支持不同算法构建的系统发育分析作为输入源。应用LSD简单三步可以快速估算tMRCA和进化速率:
Step1:获得通过ML法或其他算法构建一个系统发育树;
Step2:准备两个文件,一个采样日期(类似格式:taxa1 2001.xx)和序列的Outgroup文件;
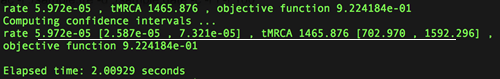
Step3:命令行运行./bin/lsd_mac -i ./sample.tre -d ./sample.date -c -r l -f 100
� �
�
如上图:估算的某植物病毒CP基因进化速率为5.972E-5 subs/site/year(95%CI: 2.587E-5-7.321E-5)
Q12: 如何应用文献报道的化石记录进行分子钟校准?
Q12: 通常两个步骤可以完成:第一,将序列导入BEAUti后,切换到“Taxon Sets” 标签,点击左下角的“+”添加一个新的sets(示例Taxa sets 名为Test),即有化石记录的Taxa归为一个Taxon Sets,并设置其他参数完毕,切换到“Prior”,双击点开参数“tmrca(Test)”的Prior窗口,选择合适的Piror Distribution(示例为Uniform),文献报道将化石校准点 Min=528.28 Ma 和 Max=636.1 Ma 分别填入 Upper 和 Lower 后的文本框内,“Generate BEAST File” 文件生成xml文件。第二,在生成的xml文件中,找到Starting Tree 位置,把冗余的代码删除,替换上自定义的Newick格式的树,如下图所示:

Q13: 如何解决设置化石校准后xml 运行过程中Prior出现 zero probability 问题?
A13:主要原因:用户自定义的起始树无法正确约束校准点,例如Q12的校准点为 528.28-636.1 Ma的 uniform prior,也就是说任何 Root age 低于528.28 Myr 或高于636.1 Ma 的树将被指定为概率为零(a zero probability). 解决办法,将 Prior Distribution 由 Uniform 改为 normalPrior,并设置合适的区间范围,将Root age 限制在528.28-636.1 Ma区间内,如下图所示:

Q14: BEAST运行程序过程中,遇到磁盘空间不够或内存不足导致xml文件末尾部分内容未运行完,如何处理?
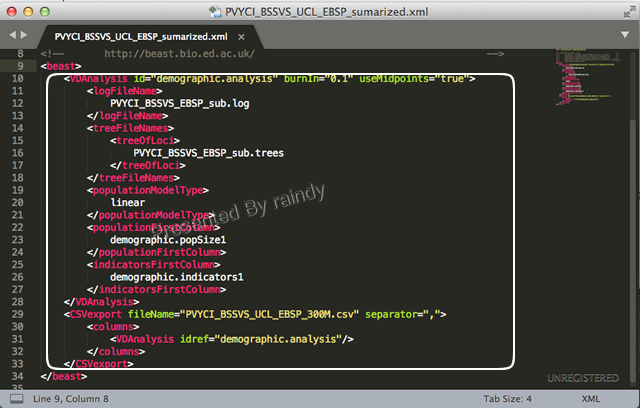
A14:近期做EBSP分析时就遇到类似的情形,除了用于EBSP plot的xx.csv 未生成外,其他分析均已完成,此时出现内存不足,重新再跑一遍是件非常痛苦的事,尤其对于大数据,有时候时间紧迫也不现实。解决的办法也简单,打开xml文件把已分析完成的部分内容删除,保留尚未分析的事项(下图所示),另存为一个xml,调用BEAST重新运行一下即可,大大节省时间。
Q15: BEAST 分析时,链长需要和采样频率设置多少比较合适?
A15:没有普适的标准,一般是10^8(8次方),最主要的一点需要保证最后的样本最少得有10,000,比如:总链长300,000,000,采样频率可以使用30,000,即保证最终样本量达到10,000。
To be continued...
https://blog.sciencenet.cn/blog-460481-1098684.html
上一篇:图解核苷酸替代模型的选择 - ModelFinder篇(简明版)
下一篇:应用 PhyloMad对进化模型准确性进行评估(一)