博文
Exploring a new ligand binding site of GPCR by MD simulation
||
Chemical Science (2018) doi: 10.1039/C8SC01680A
Identifying a target ligand binding site is an important step for structure-based rational drug design as shown here for G protein-coupled receptors (GPCRs), which are among the most popular drug targets. We applied long-time scale molecular dynamics simulations, coupled with mutagenesis studies, to two prototypical GPCRs, the M3 and M4 muscarinic acetylcho-line receptors. Our results indicate that unlike the synthetic antagonists, which bind to the classic orthosteric site, the endogenous agonist acetylcholine is able to diffuse into a much deeper binding pocket. We also discovered that the most recently resolved crystal structure of LTB4 receptor comprised a bound inverse agonist, which extended its benzamidine moiety to the same binding pocket discovered in this work. Analysis on all resolve GPCR crystal structures indicated that this new pocket could exist in most receptors. Our findings provide new opportunities for GPCR drug discovery.
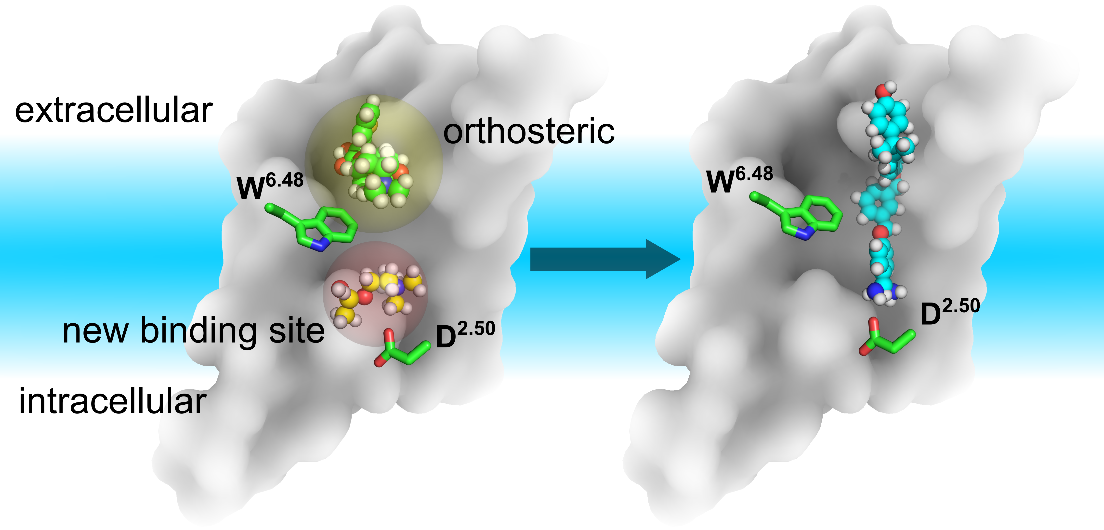
The relative positions of the classic orthosteric site (yellow shaded sphere) and the newly discovered novel ligand binding site(left panel, red shaded sphere) located between W6.48 an D2.50. A new drug molecule (right panel) can be designed, based on the newly discovered site in this work. Green balls-and-sticks: TTP molecule observed in the crystal structures of M3 & M4 receptors. Yellow balls-and-sticks: ACh observed in this work. Cyan stick: an inverse agonist observed in the crystal structure of LTB4 receptor.
https://blog.sciencenet.cn/blog-355217-1124563.html
上一篇:First GPCR-directed antibody passes approval milestone
下一篇:The first Frizzled class GPCR structure was resolved