博文
Computational modelling identified the real catalytic site
||
GyrI-like proteins catalyze cyclopropanoid hydrolysis to confer cellular protection. Nature Communications, Yuan H., Zhang J., Cai Y., Wu S., Yang K., Chan S., Huang W., Jin W., Li Y., Yin Y., Igarashi Y., Yuan S.*, Zhou J.*, Tang G.* (2017), doi:10.1038/s41467-017-01508-1
Mutagenesis has been widely applied to enzyme catalysis and structural biology. To understand how an enzyme catalyzes a specific substrate, biologists usually mutate residue(s) in the vicinity of the catalytic site, in the hope to obtain the complex structure of enzyme-substrate.
However, the mutagenesis might introduce artifacts: the changes of binding pocket could lead the substrate to bind to somewhere else. Sometimes, the position of a substrate observed in the crystal structure of mutated enzymes could be far away from the actual catalytic site.
In our recent work (Nature Communications, 2017, doi:10.1038/s41467-017-01508-1), we observed this limitation of structural biology in enzyme CCHs. Mutagenesis following the binding mode observed in crystal structure doesn’t show too much effect on CCHs’ activity. Moreover, no any nucleophile residues were observed next to the substrate in the crystal structure. To resolve this puzzle, we introduced molecular docking, coupled with molecular dynamics simulation, to identify the real binding mode of this enzyme. The subsequent mutagenesis studies confirmed our predicted new binding mode of the substrate.
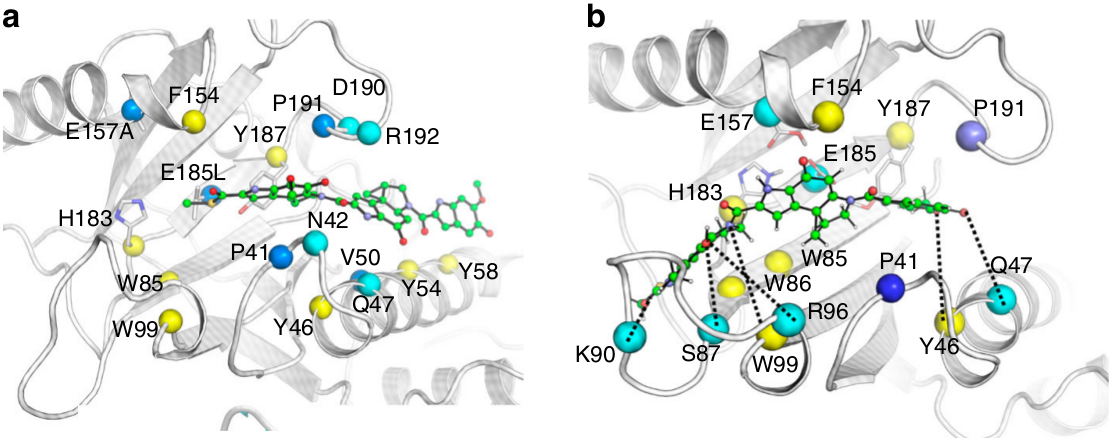
Figure 1. The binding modes of the substrate in CCHs. (a) The binding mode observed in crystal structure. (b) The binding mode predicted by computational modelling.
https://blog.sciencenet.cn/blog-355217-1085550.html
上一篇:A postodoc position in EPFL, Swtizerland
下一篇:Minimizing all windows