博文
重磅!哈佛大学李恒团队提出一种全新的单倍型基因组组装算法hifiasm
||
北京时间2021年2月2日凌晨0时,美国哈佛大学医学院Dana-Farber癌症研究所李恒课题组在Nature Methods杂志发表论文“Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm”。
该研究提出一种全新的单倍型基因组组装算法hifiasm,能够有效地对大型复杂基因组生成高质量的单倍型组装结果。
李恒教授为论文的通讯作者,程昊宇博士为第一作者。
单倍型组装的难点
单倍型基因组组装是研究基因组结构与变异的最理想方式。由于技术的局限,大多数组装算法倾向于将不同单倍型有损的压缩成一条混合的代表性序列。对于自然界中主流的二倍体和多倍体样本而言,这类方法损失了大量的单倍型信息。这使得长久以来,研究人员难以对高杂合、高重复的基因组进行深入的分析。
为了解决这个难题,一些组装算法首先生成混合的代表性序列,接着从代表性序列中恢复出不同的单倍型信息。但是,由于代表性序列本身已经丢失了大量的信息,这类方法难以获得高质量的单倍型组装结果。
近期的组装算法通过额外的信息,如家系(Trio binning)或者Hi-C等数据,预先全局性的将待组装的测序读段划分到不同单倍型,再进行分别组装,从而试图获得高质量的单倍型组装结果。但是对于低杂合的样本而言,这种方法难以做到完美的预先划分,因此容易产生组装错误。
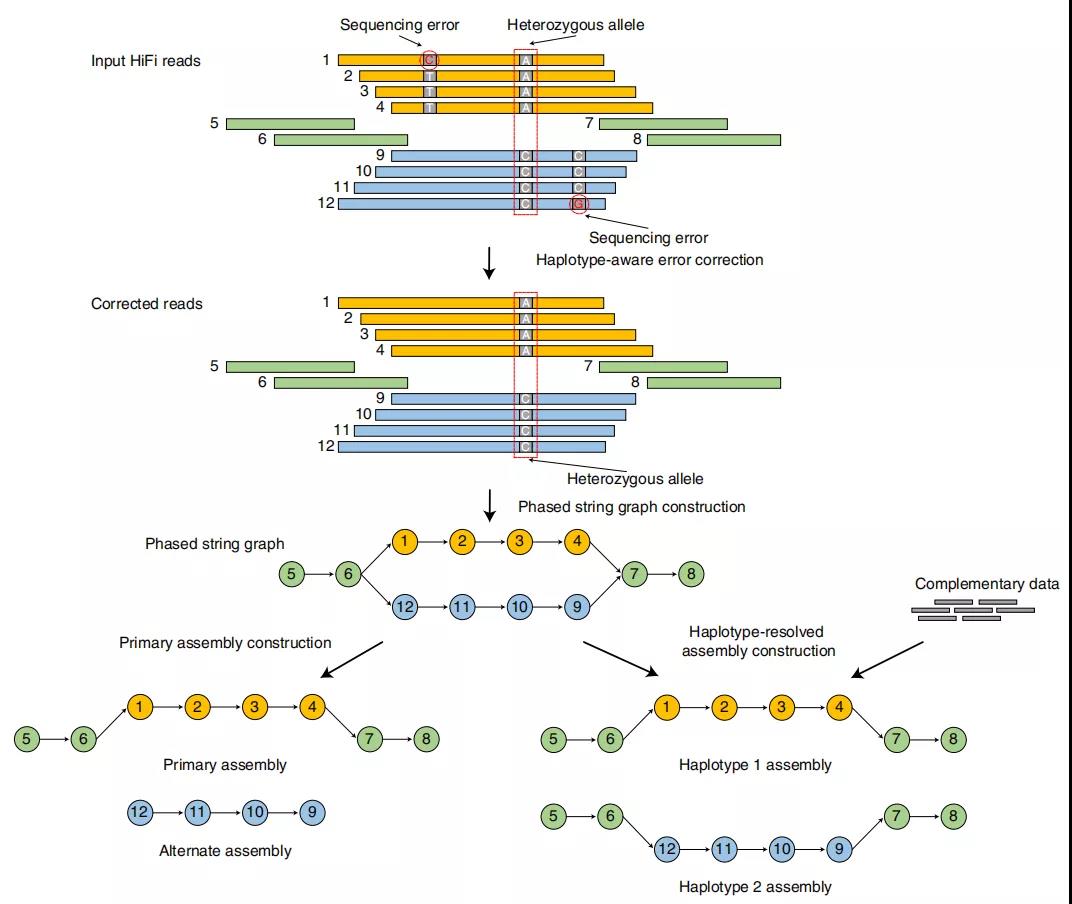
图1. hifiasm算法流程
Hifiasm算法
在本研究中,研究人员提出了一种全新的针对PacBio HiFi (High-Fidelity reads) 数据的单倍型组装算法hifiasm。该算法有两项重要创新。
第一,提出了单倍型敏感的组装思路,使得在组装的全过程中能够无损的保留单倍型信息,同时也极大的提升了对基因组高重复和复杂区域的解析能力。
第二,提出了Graph-binning的分型策略,其利用组装图的结构信息对全局分型结果进行校正,从而极大地提高了单倍型组装的质量。Graph-binning不对待组装的测序读段进行预先全局划分,因此能够克服划分错误带来组装问题。
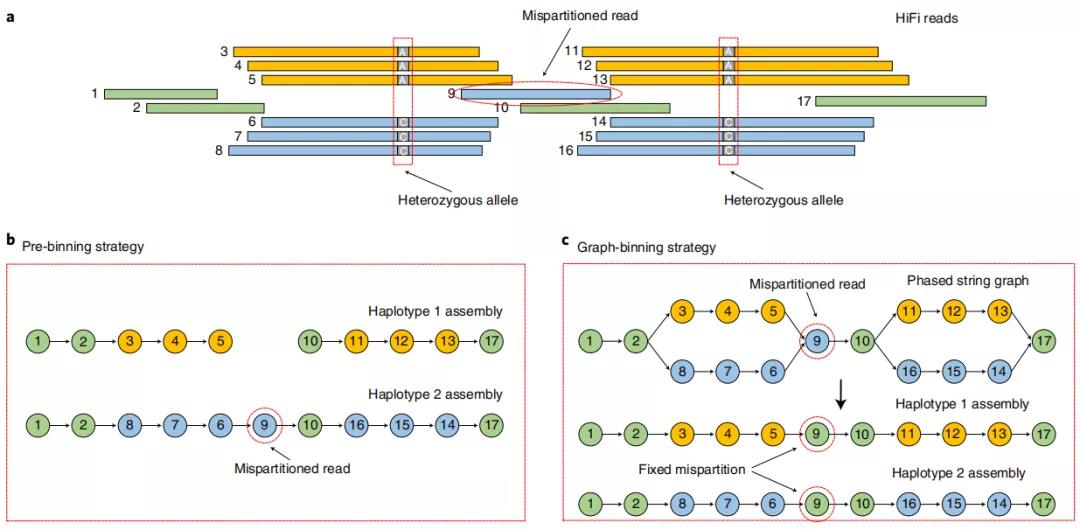
图2. Graph-binning分型策略
组装结果
研究人员在不同的数据集上测试了hifiasm算法。对于不同大小,不同杂合度和不同单倍型数量的动物和植物基因组,hifiasm能够产生质量最高的组装结果。尤其值得注意的是,hifiasm仅用三天时间,就完成27Gb超大加州红杉基因组的组装,并且组装结果的连续性7倍于其他算法。
对于人类基因组,hifiasm也能取得最好的效果。相比于现有算法,hifiasm所产生的组装结果连续性最高,同时也正确解析了最多的复杂和高重复区域,如MHC (主要组织相容性复合体)和centromere (着丝端粒)。尤其对于人类二倍体样本HG002和HG00733,hifiasm产生的组装结果的连续性3倍于其他算法,并成功保留了最多的变异信息。
未来展望
目前,hifiasm算法已经在诸如Human Pangenome Project (人类泛基因组计划) 等项目中被广泛使用。预计在不久的将来,单倍型基因组组装将成为人类和动植物研究中的通用模式。
相关论文信息:
https://doi.org/10.1038/s41592-020-01056-5
https://blog.sciencenet.cn/blog-3423233-1270140.html
上一篇:哈佛医学院团队揭示人类神经元如何编码对他人的看法
下一篇:上海师范大学黄学辉团队开发水稻QTL基因变异图谱导航系统