博文
基于单个模板的乳酸脱氢酶进行建模--Modeller基础教程
|||
写在前面,modeller软件的安装可以参考官网,我这里使用的Ubuntu系统,这个例子的数据文件可以从官网下载。
1、将蛋白质序列输入文件配置为modeller的文件格式
首先,必须将目标蛋白质序列设置为可由MODELLER读取的PIR格式(文件后缀'.ali')
>P1;TvLDH
sequence:TvLDH:::::::0.00: 0.00
MSEAAHVLITGAAGQIGYILSHWIASGELYGDRQVYLHLLDIPPAMNRLTALTMELEDCAFPHLAGFVATTDPKA
AFKDIDCAFLVASMPLKPGQVRADLISSNSVIFKNTGEYLSKWAKPSVKVLVIGNPDNTNCEIAMLHAKNLKPEN
FSSLSMLDQNRAYYEVASKLGVDVKDVHDIIVWGNHGESMVADLTQATFTKEGKTQKVVDVLDHDYVFDTFFKKI
GHRAWDILEHRGFTSAASPTKAAIQHMKAWLFGTAPGEVLSMGIPVPEGNPYGIKPGVVFSFPCNVDKEGKIHVV
EGFKVNDWLREKLDFTEKDLFHEKEIALNHLAQGG*
第一行包含序列代码,格式为“> P1; code”。如果有必要的话,第二行包含用冒号分隔的十个字段,通常包含有关结构文件的信息。这些字段中只有两个用于序列,“序列”(指示文件包含一个没有已知结构的序列)和“ TvLDH”(模型文件名)。文件的其余部分包含TvLDH的蛋白质序列,并带有“ *”标记其结尾。使用标准的单字母氨基酸代码。(请注意,它们必须是大写字母;一些小写字母用于非标准残基。有关更多信息,请参阅Modeller发行版中的modlib / restyp.lib文件。)
2、选择与目标蛋白质相似度高的已知结构的PDB序列
我们使用'build_profile.py'脚本进行筛选与目标序列相似度较高的已知结构的PDB序列,
"build_profile.py"脚本如下:
from modeller import *
log.verbose()
env = environ()
#-- Prepare the input files
#-- Read in the sequence database
sdb = sequence_db(env)
sdb.read(seq_database_file='pdb_95.pir', seq_database_format='PIR',
chains_list='ALL', minmax_db_seq_len=(30, 4000), clean_sequences=True)
#-- Write the sequence database in binary form
sdb.write(seq_database_file='pdb_95.bin', seq_database_format='BINARY',
chains_list='ALL')
#-- Now, read in the binary database
sdb.read(seq_database_file='pdb_95.bin', seq_database_format='BINARY',
chains_list='ALL')
#-- Read in the target sequence/alignment
aln = alignment(env)
aln.append(file='TvLDH.ali', alignment_format='PIR', align_codes='ALL')
#-- Convert the input sequence/alignment into
# profile format
prf = aln.to_profile()
#-- Scan sequence database to pick up homologous sequences
prf.build(sdb, matrix_offset=-450, rr_file='${LIB}/blosum62.sim.mat',
gap_penalties_1d=(-500, -50), n_prof_iterations=1,
check_profile=False, max_aln_evalue=0.01)
#-- Write out the profile in text format
prf.write(file='build_profile.prf', profile_format='TEXT')
#-- Convert the profile back to alignment format
aln = prf.to_alignment()
#-- Write out the alignment file
aln.write(file='build_profile.ali', alignment_format='PIR')
1、通过创建一个新的“环境”对象来初始化此建模运行的“环境”。几乎所有的MODELLER脚本都需要执行此步骤,因为构建大多数其他有用的对象都需要新对象(在这里我们称其为“ env”,也可以随意命名)。
2、创建一个新的“ sequence_db”对象,将其称为“ sdb”。'sequence_db'对象用于包含蛋白质序列的大型数据库。
3、将包含非冗余PDB序列(具有95%序列同一性)的文本格式文件读入sdb数据库。序列可在文件“ pdb_95.pir”中找到(可使用此页面顶部的链接下载)。与以前创建的对齐方式一样,此文件为PIR格式。丢弃少于30个或超过4000个残基的序列,并去除非标准残基。
4、写入二进制机器专用文件,其中包含在上一步中读取的所有序列。
5、重新读回二进制格式文件。请注意,如果您打算多次使用同一数据库,则应该仅在第一次使用前两个步骤来生成二进制数据库。在随后的运行中,您可以忽略这两个步骤,而直接使用二进制文件,因为读取二进制文件比读取PIR文件要快得多。
6、创建一个名为“ aln”的新“ alignment”对象,从文件“ TvLDH.ali”中读取查询序列“ TvLDH”,并将其转换为配置文件“ prf”。配置文件包含与比对相似的信息,但更紧凑,更适合序列数据库搜索。
7、在序列数据库“ sdb”中搜索我们的查询配置文件“ prf”。来自序列数据库的匹配项将添加到配置文件中。
8、写入查询序列及其同源文件的配置文件(请参见文件“ build_profile.prf”)。等效信息也以标准对齐格式写出。
执行命令:
python2 build_profile.py > build_profile.log
输出文件:"build_profile.ali","build_profile.prf",用于后续的比对使用。
"build_profile.prf"文件的内容如下:
# Number of sequences: 30
# Length of profile : 335
# N_PROF_ITERATIONS : 1
# GAP_PENALTIES_1D : -900.0 -50.0
# MATRIX_OFFSET : 0.0
# RR_FILE : ${MODINSTALL8v1}/modlib//as1.sim.mat
1 TvLDH S 0 335 1 335 0 0 0 0. 0.0
2 1a5z X 1 312 75 242 63 229 164 28. 0.83E-08
3 1b8pA X 1 327 7 331 6 325 316 42. 0.0
4 1bdmA X 1 318 1 325 1 310 309 45. 0.0
5 1t2dA X 1 315 5 256 4 250 238 25. 0.66E-04
6 1civA X 1 374 6 334 33 358 325 35. 0.0
7 2cmd X 1 312 7 320 3 303 289 27. 0.16E-05
8 1o6zA X 1 303 7 320 3 287 278 26. 0.27E-05
9 1ur5A X 1 299 13 191 9 171 158 31. 0.25E-02
10 1guzA X 1 305 13 301 8 280 265 25. 0.28E-08
11 1gv0A X 1 301 13 323 8 289 274 26. 0.28E-04
12 1hyeA X 1 307 7 191 3 183 173 29. 0.14E-07
13 1i0zA X 1 332 85 300 94 304 207 25. 0.66E-05
14 1i10A X 1 331 85 295 93 298 196 26. 0.86E-05
15 1ldnA X 1 316 78 298 73 301 214 26. 0.19E-03
16 6ldh X 1 329 47 301 56 302 244 23. 0.17E-02
17 2ldx X 1 331 66 306 67 306 227 26. 0.25E-04
18 5ldh X 1 333 85 300 94 304 207 26. 0.30E-05
19 9ldtA X 1 331 85 301 93 304 207 26. 0.10E-05
20 1llc X 1 321 64 239 53 234 164 26. 0.20E-03
21 1lldA X 1 313 13 242 9 233 216 31. 0.31E-07
22 5mdhA X 1 333 2 332 1 331 328 44. 0.0
23 7mdhA X 1 351 6 334 14 339 325 34. 0.0
24 1mldA X 1 313 5 198 1 189 183 26. 0.13E-05
25 1oc4A X 1 315 5 191 4 186 174 28. 0.18E-04
26 1ojuA X 1 294 78 320 68 285 218 28. 0.43E-05
27 1pzgA X 1 327 74 191 71 190 114 30. 0.16E-06
28 1smkA X 1 313 7 202 4 198 188 34. 0.0
29 1sovA X 1 316 81 256 76 248 160 27. 0.93E-03
30 1y6jA X 1 289 77 191 58 167 109 33. 0.32E-05
根据"build_profile.py"脚本的输出信息,我们主要看第二、十、十一、十二列的信息,第二列是与目标序列比对上的PDB编号,第十一行是序列相似度(identity),第十二行是e-value值。这里我们的选择标准是首先选择e-value值越小越好,序列相似度越大越好,挑选4-10个已知结构的蛋白质序列。如预期的那样,所有命中对应于苹果酸脱氢酶(1bdm:A,5mdh:A,1b8p:A,1civ:A,7mdh:A和1smk:A)。选出相似度高的蛋白质序列后,我们使用compare.py再从这6个结构当中选择最合适的作为模板。
compare.py脚本如下:
from modeller import *
env = environ()
aln = alignment(env)
for (pdb, chain) in (('1b8p', 'A'), ('1bdm', 'A'), ('1civ', 'A'),
('5mdh', 'A'), ('7mdh', 'A'), ('1smk', 'A')):
m = model(env, file=pdb, model_segment=('FIRST:'+chain, 'LAST:'+chain))
aln.append_model(m, atom_files=pdb, align_codes=pdb+chain)
aln.malign()
aln.malign3d()
aln.compare_structures()
aln.id_table(matrix_file='family.mat')
env.dendrogram(matrix_file='family.mat', cluster_cut=-1.0)
这个文件我们在做不同的蛋白质结构预测时需要进行相应的配置。在运行这个脚本之前我们需要把相应的PDB序列文件下载到当前工作目录下。(请注意,为了使此功能起作用,您必须将所有PDB文件都与此脚本放在同一目录中,该文件可以从PDB网站下载。)
脚本执行命令:
python2 compare.py > compare.log
得到的"compare.log"文件内容如下:
Sequence identity comparison (ID_TABLE):
Diagonal ... number of residues;
Upper triangle ... number of identical residues;
Lower triangle ... % sequence identity, id/min(length).
1b8pA @11bdmA @11civA @25mdhA @27mdhA @21smkA @2
1b8pA @1 327 194 147 151 153 49
1bdmA @1 61 318 152 167 155 56
1civA @2 45 48 374 139 304 53
5mdhA @2 46 53 42 333 139 57
7mdhA @2 47 49 87 42 351 48
1smkA @2 16 18 17 18 15 313
Weighted pair-group average clustering based on a distance matrix:
.----------------------- 1b8pA @1.9 39.0000
|
.-------------------------------- 1bdmA @1.8 50.5000
|
.------------------------------------ 5mdhA @2.4 55.3750
|
| .--- 1civA @2.8 13.0000
| |
.---------------------------------------------------------- 7mdhA @2.4 83.2500
|
.------------------------------------------------------------ 1smkA @2.5
+----+----+----+----+----+----+----+----+----+----+----+----+
86.0600 73.4150 60.7700 48.1250 35.4800 22.8350 10.1900
79.7375 67.0925 54.4475 41.8025 29.1575 16.5125
这里一共聚了三类:第一类有三个(5mdh:A,1bdm:A和1b8p:A),第二类有两个(1civ:A和7mdh:A),第三类有一个(1smk:A)。上面的比较显示1civ:A和7mdh:A在顺序和结构上几乎相同。但是,7mdh:A具有更好的晶体分辨率(2.4A对2.8A),消除了1civ:A。第二组结构(5mdh:A,1bdm:A和1b8p:A)具有一些相似之处。在这一组中,5mdh:A的分辨率最差,仅考虑1bdm:A和1b8p:A。1smk:A是整个可能模板集合中最多样化的结构。但是,它与查询序列的序列同一性最低(34%)。我们最终选择1bdm:A而不是1b8p:A和7mdh:A,因为它具有更好的结晶R因子(16.9%)和更高的整体序列与查询序列同一性(45%)。
3、将目标蛋白质序列与挑选出来的模板序列进行比对
利用"align2d.py"脚本进行比对,该脚本如下:
from modeller import *
env = environ()
aln = alignment(env)
mdl = model(env, file='1bdm', model_segment=('FIRST:A','LAST:A'))
aln.append_model(mdl, align_codes='1bdmA', atom_files='1bdm.pdb')
aln.append(file='TvLDH.ali', align_codes='TvLDH')
aln.align2d()
aln.write(file='TvLDH-1bdmA.ali', alignment_format='PIR')
aln.write(file='TvLDH-1bdmA.pap', alignment_format='PAP')
脚本运行命令为:
python2 align2d.py > align2d.log
该脚本运行得到"TvLDH-1bdmA.ali","TvLDH-1bdmA.pap"两个文件,用于后续的模型构建。
4、模型构建
根据比对的结果进行同源结构建模,一旦构建了目标模板对齐方式,MODELLER就会使用其自动模型类自动完全计算目标的3D模型。以下脚本将基于1bdm生成五个类似的TvLDH模型:模板结构和文件“ TvLDH-1bdmA.ali”(脚本“ model-single.py”)中的对齐方式。
脚本“ model-single.py”如下:
from modeller import *
from modeller.automodel import *
#from modeller import soap_protein_od
env = environ()
a = automodel(env, alnfile='TvLDH-1bdmA.ali',
knowns='1bdmA', sequence='TvLDH',
assess_methods=(assess.DOPE,
#soap_protein_od.Scorer(),
assess.GA341))
a.starting_model = 1
a.ending_model = 5
a.make()
执行命令:
python2 model-single.py > model-single.log
最重要的输出文件是“ model-single.log”,该文件报告警告,错误和其他有用的信息,包括用于建模的输入限制,这些限制在最终模型中被违反。该日志文件的最后几行如下所示。
>> Summary of successfully produced models:
Filename molpdf DOPE score GA341 score
----------------------------------------------------------------------
TvLDH.B99990001.pdb 1763.56104 -38079.76172 1.00000
TvLDH.B99990002.pdb 1560.93396 -38515.98047 1.00000
TvLDH.B99990003.pdb 1712.44104 -37984.30859 1.00000
TvLDH.B99990004.pdb 1720.70801 -37869.91406 1.00000
TvLDH.B99990005.pdb 1840.91772 -38052.00781 1.00000
该日志文件提供了所有已构建模型的摘要。对于每个模型,它都会列出文件名,其中包含PDB格式的模型坐标。可以通过读取PDB格式的任何程序(例如Chimera)查看模型。日志还显示了每个模型的得分,下面将进一步讨论。(请注意,您的计算机上的实际数字可能会略有不同-无需担心。)
可以在https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html打开TvLDH.B99990001.pdb
5、模型评估
如果针对同一目标计算了多个模型,则可以通过以下几种方式选择“最佳”模型。例如,您可以选择具有最低MODELLER目标函数值或"DOPE"或"SOAP score"或具有最高"GA341 score"的模型,这些数据在上面的日志文件末尾报告(目标函数molpdf总是会被计算,并且还会在每个生成的PDB文件的REMARK中报告。仅当您将它们列在评估方法中时,才会计算DOPE,SOAP和GA341分数或任何其他评估分数。要计算SOAP分数,您首先需要从SOAP网站下载SOAP-Protein潜在文件,然后通过删除'#'字符取消对model-single.py中与SOAP相关的行的注释。)。molpdf,DOPE和SOAP分数不是“绝对”的量度,因为它们只能用于对根据相同比对计算出的模型进行排名。其他分数可以转让。例如,GA341分数始终在0.0(最差值)到1.0(原生值)之间变化;但是,GA341在区分“好”模型和“坏”模型方面不如DOPE或SOAP好。
没有模型评估分数是完美的,因此您不应该期望它们具有完美的相关性。“好”模型的GA341得分接近1.0(越大越好),但DOPE得分越小(DOPE得分(如molpdf)是“动能”,因此较小或为负更好)。
无论是DOPE还是GA341都没有表现出很多对劣质模型的歧视-例如您不能真正说出GA341得分为0.1的模型比得分为0.2的模型“更差”(开发该得分是为了区分好模型和坏模型,而不是对坏模型进行排名)。任何比0.6差的模型都是不好的模型。
在对模型进行任何外部评估之前,应检查建模运行中的日志文件是否存在运行时错误(“ model-single.log”)和限制违反情况(有关详细信息,请参阅《 MODELLER手册》)。脚本“ evaluate_model.py”评估具有DOPE潜力的输入模型。(请注意,这里我们任意选择了第二个生成的模型-您可能想要尝试其他模型。)
利用脚本“ evaluate_model.py”和“ evaluate_template.py”对构建的蛋白质结构和模板蛋白质结构进行评价;看看那些区域是合理的,那些区域是不合理的。
"evaluate_model.py"脚本如下:
from modeller import * from modeller.scripts import complete_pdb log.verbose() # request verbose output env = environ() env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters # read model file mdl = complete_pdb(env, 'TvLDH.B99990002.pdb') # Assess with DOPE: s = selection(mdl) # all atom selection s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='TvLDH.profile', normalize_profile=True, smoothing_window=15)
运行该脚本命令:
python2 evaluate_model.py >evaluate_model.log
该脚本会产生"TvLDH.profile"文件,可以用于后续的作图。
"evaluate_template.py"脚本如下:
from modeller import *
from modeller.scripts import complete_pdb
log.verbose() # request verbose output
env = environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology
env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters
# directories for input atom files
env.io.atom_files_directory = './:../atom_files'
# read model file
mdl = complete_pdb(env, '1bdm.pdb', model_segment=('FIRST:A', 'LAST:A'))
s = selection(mdl)
s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='1bdmA.profile',
normalize_profile=True, smoothing_window=15)
运行该脚本命令:
python2 evaluate_template.py >evaluate_template.log
该脚本会产生"1bdmA.profile"文件,可以用于后续的作图。
使用"plot_profile.py"脚本可以根据"TvLDH.profile"和"1bdmA.profile"文件进行作图,该脚本运行后会产生一个氨基酸的能量折线图,执行命令如下:
python2 plot_profile.py > plot_profile.log
图形如下:
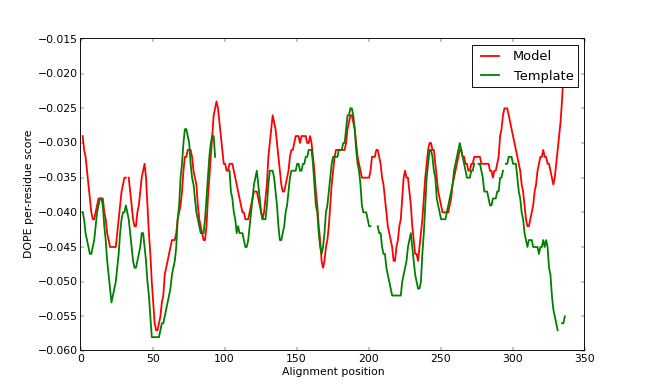
GA341得分以及PROCHECK程序的外部分析证实TvLDH.B99990001.pdb是合理的模型。但是,绘制的DOPE得分曲线(上图)显示了残基90和100之间的长活性位点环与靶序列C末端的长螺旋之间相对较高的能量区域。(请注意,我们已将模型轮廓叠加在模板轮廓上-可以看到图中的间隙与路线中的间隙相对应。请记住,分数不是绝对的,因此我们无法在两者之间进行直接的数值比较。我们可以通过比较两个轮廓的粗略形状来了解输入对齐方式的质量,如果一个轮廓相对于另一个轮廓明显偏移,则对齐方式也有可能偏离了正确的对齐方式。)
该长环与区域220-250相互作用,该区域形成了活动部位的另一半。后一部分在模板中得到很好的解析,并可能在目标结构中正确建模,但是由于与90-100区域的不利的非键交互作用,DOPE也报告说它具有很高的“能量”。通常,DOPE指示的可能错误不一定是实际错误,尤其是如果它突出显示了一个活性位点或一个蛋白质-蛋白质界面时。但是,在这种情况下,相同的活动站点环在模板结构中具有更好的配置文件,这加强了对模型在活动站点区域中可能不正确的评估。高级建模教程中解决了此问题。
参考:
1、https://salilab.org/modeller/tutorial/basic.html
2、
https://blog.sciencenet.cn/blog-3416913-1225133.html
上一篇:批量下载某一物种的基因序列
下一篇:antismash 本地安装教程