博文
采用Stacks2分析RADseq数据[无参]
|||
Stacks2的安装:
Stacks目前最新版是2.1版。在安装前需用以下命令确认一下电脑上的gcc版本,因为Stacks2.1需要gcc 4.9.0及以上编译器。
gcc --version
如果gcc版本过低时可用以下方式升级:
参见http://ftp.gnu.org/gnu/gcc/,可以找到想要下载的gcc压缩包,以gcc4.9.4为例:
#建立工作目录stacks2
mkdir stacks2 && cd stacks2
# 下载gcc4.9.4压缩包和stacks2.1压缩包
wget http://ftp.gnu.org/gnu/gcc/gcc-4.9.4/gcc-4.9.4.tar.gz
wget http://catchenlab.life.illinois.edu/stacks/source/stacks-2.1.tar.gz
# 先更新gcc
tar -zxvf gcc-4.9.4.tar.gz
cd gcc-4.9.4/
./contrib/download_prerequisites
# 这里建了两个文件夹,一个是build用于安装,一个是gccbin用于存放最终的结果
注意:一定要用--prefix指定gccbin作为最终存放二进制文件的目录,要不然会使用默认的/usr/bin,make的时候会因没有权限而报错!
mkdir build gccbin && cd build
../configure --prefix=/path/to/gcc-4.9.4/gccbin -enable-checking=release -enable-languages=c,c++ -disable-multilib
# /path/to/gcc-4.9.4/gccbin表示gccbin目录的位置,建议用绝对路径!
#因为编译时间较久,建议用多线程make,这里的-j指定线程数
make -j12
make install
# 将gccbin下的bin目录添加到PATH中,将gccbin下的lib64目录添加到LD_LIBRARY_PATH中。
注意:这一步一定要添加,特别是LD_LIBRARY_PATH!如果想长期使用这个版本的gcc,还可以将其添加到$HOME/.bashrc文件中并source
export PATH=/path/to/gcc-4.9.4/gccbin/bin:$PATH
export LD_LIBRARY_PATH=/path/to/gcc-4.9.4/gccbin/bin/lib64/:$LD_LIBRARY_PATH
到此gcc升级已完成,可通过gcc --version查看是否成功升级,如果出现以下结果则表示成功。
tar -zxvf stacks-2.1.tar.gz
cd stacks-2.1
./configure --prefix=$PWD
make && make install
可以将stacks路径添加到系统路径中,如果长期使用可添加到$HOME/.bashrc
export PATH=/path/to/stacks-2.1/bin:$PATH
完成后在stacks-2.1目录下运行bin/ustacks,如果出现以下信息,则表示安装成功
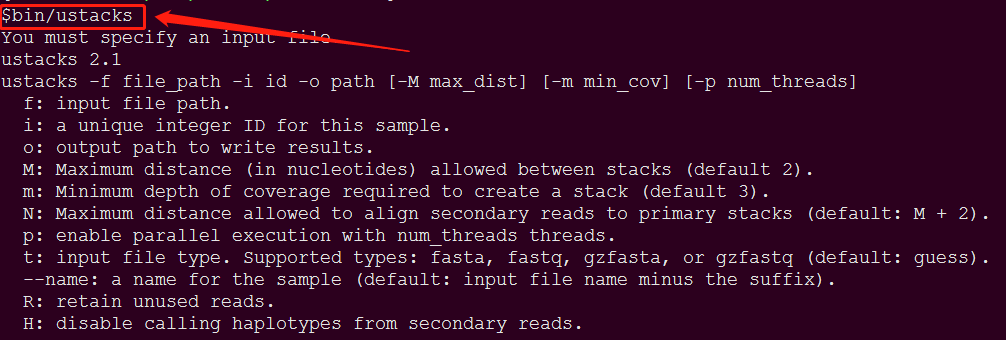
注意:如果出现以下libstdc++.so.6错误信息,表明gcc路径不对,且LD_LIBRARY_PATH的路径也没添加上来!

无参时,Stacks2可以用denovo_map.pl流程完成所有的分析内容。
输入:
PE fastq.gz文件,map_file.pop文件。
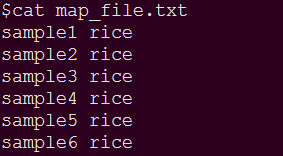
map_file.pop文件格式为:样本名[TAB]组名。如果没有群体分组信息,则所有样本的组名写成相同的名字,具体如下:
一步到位:
如果输入的是gz压缩的PE reads具体代码如下:
denovo_map.pl --samples 01.fastq/ --popmap map_file.pop -o stack_output/ -T 24 --paired
其中所有reads均存放在01.fastq目录下,并且必须命令为<ID>.1.fq.gz和<ID>.2.fq.gz,<ID>为样本id,例如sample1.1.fq.gz和sample2.2.fq.gz。并且fastq头行必须要有/1标识Reads 1,/2标识Reads 2,如下Reads1和Reads2中的头行


注意:stacks分析时所有样本的reads长度必须一致!
分步完成:
1. 前处理
如果fastq头行没有/1和/2标识,可采用Stack2自带的process_radtags完成前处理。当然,这个程序能做的事远不止转个格式,它还可以去除低质量的序列,demuliplexing,去除barcode等。
这里以样本sample1为例
mkdir sample1
process_radtags -1 sample1.R1.fastq.gz -2 sample1.R2.fastq.gz -q -c --merge -i gzfastq --disable_rad_check -o sample1
rename sample_unbarcoded sample1 sample1/*.gz
#完成后会在Sample1目录下生成以下文件
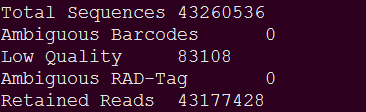
(1) process_radtags.01.RawData.log
这个文件会记录下总的有多少条reads,因什么原因去掉reads,去掉了多少等信息,如下
(2) sample1.1.fq.gz和sample1.2.fq.gz
这两个文件是过滤后可用于后续分析的文件
(3) sample1.rem.1.fq.gz和sample1.rem.2.fq.gz
这两个文件是质量低被弃除的reads
为了完成reads的组装(聚类),stacks采用两步法[1]:
(1) 用ustacks分别对单个样本的R1进行组装(聚类),获取consensus序列。这一步生成的结果称为loci;
(2) 再使用cstacks将所有样本的loci聚合到一起。这一步生成的结果称为catalog。
在ustacks中控制组装结果的最关键参数是M,它表示在一个杂合样本中两个等位基因容许的mismatch数,默认为2;在cstacks中控制组装结果的最关键参数是n,它表示群体间等位基因容许的mismatch数,默认为1。这两个参数设置时差异不要过大,建议设置为相同的值[2]。具体的选取可采用RADassembly软件[见文献3,后续会有相应博文],另外Stacks官网上还推荐了两种方法,分别见文献[4-5]。
2. ustacks
ustacks -t gzfastq -f sample1.1.fq.gz -o stack_output -i 1 --name sample1 -p 24
注意:在这里有个-i选项,它是整套分析流程中样本的ID,需为整数类型。
# 采用最大似然模型对每个样本单独组装(个人觉得叫做“聚类”会更贴切),顺带给SNP calling。这步运行完成后每个样本生成三个文件,以sample1为例:sample1.tags.tsv.gz,sample1.snps.tsv.gz和sample1.alleles.tsv.gz。另外,在log文件中会记录下每个样本的深度情况。
tags.tsv用于存入assembled loci序列信息,各列解释:
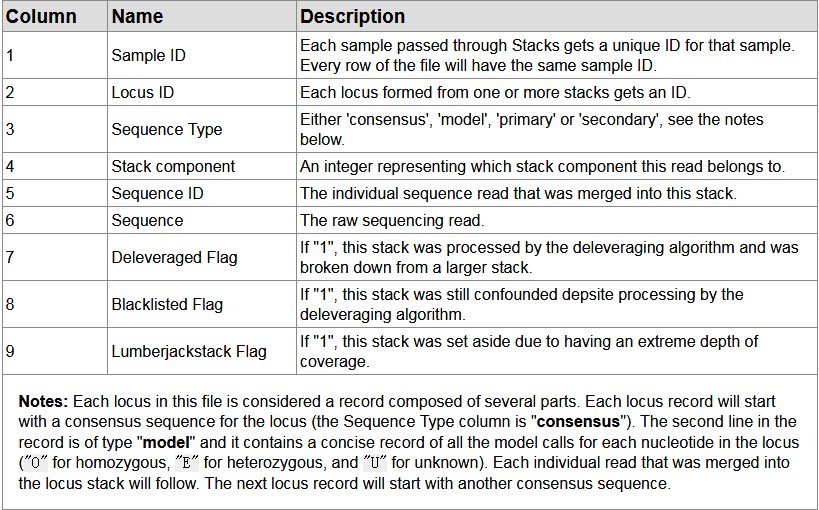
snps.tsv: Model calls from each locus,各列解释:

alleles.tsv: Haplotypes/alleles recorded from each locus,各列解释:

3. cstacks
cstacks -P stack_output -M map_file.pop -n 4 -p 24
#cstacks采用kmer的办法来进行比对。ustacks生成的三个文件在这一步都会用到。这步运行完所有样本合在一起生成三个文件:catalog.allels.tsv.gz, catalog.snps.tsv.gz和catalog.tags.tsv.gz。其中最重要的是catalog.tags.tsv.gz,它是所有loci合在一起组装成的catalog,相当一个超级consensus文件。
ustacks的tags.tsv.gz文件中除记录有consensus序列外还有model和primary,但是cstacks的tags.tsv.gz文件中仅有consensus序列。其各列解释与ustacks文件中生成的对应文件相同。
4. sstacks
sstacks -P ./stack_output/ -p 8 -M map_file.pop
# 上一步cstacks生成的三个文件,这步均会用到。sstack将各样本的PEreads中R1序列match到上步cstacks生成的catalog上,运行完生成每个样本的matches.tsv.gz文件,具体如下:
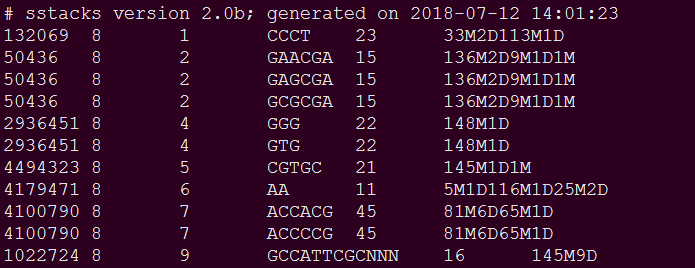
matches.tsv: Matches to the catalog,各列解释:

5. tsv2bam
tsv2bam -P ./stack_output/ -M map_file.pop -t 8 -R /lustre/Work/project/resequencing/20180514_17141_xun/09.stacks/91.fastq
# tsv2bam将sstacks生成的各样本tsv文件转为各locus的bam文件。运行完后,每个样本生成一个matchs.bam文件。其中Sample ID即ustacks中-i选项指定的样本ID
注:到这一步为止,以上各步,仅采用R1 reads。R2 reads在下一步才会用到
6. gstacks
gstacks -P stack_output/ -M map_file.pop -t 24
# gstacks主要用于merge PE reads成contig,然后将reads比对到locus上,并在群体水平call 变异以及分型。这步运行完生成catalog.fa.gz和catalog.calls,其中catalog.calls是个二进制文件。同时还生成了一个gstacks.log.distribs文件虽然给出了诸如“sample , n_loci, n_used_fw_reads mean_cov, mean_cov_ns"这样的表头信息,但是没有解释,具体不详。
7. populations
populations --in_path stack_output/ --popmap map_file.pop --threads10 --fasta_samples --fasta_loci --vcf
# populations程序在群体水平进行汇总统计(如计算Fst等),运行完生成以下文件:
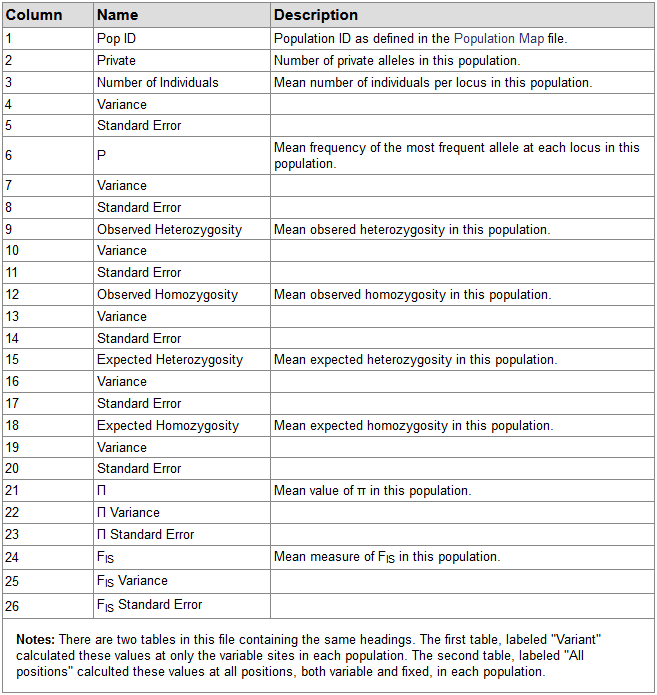
populations.sumstats.tsv: Summary statistics for each population,各列解释:

populations.sumstats_summary.tsv: Summary of summary statistics for each population,各列解释:
populations.samples.fa : Per-locus, per-haplotype sequences
如果指定--fasta_samples,才会生成该文件,该文件提供了全部RAD locus序列,这个文件和populations.haplotypes.tsv相对应。
例如:
>CLocus_10056_Sample_934_Locus_12529_Allele_0 [sample_934; groupI, 49712]
TGCAGGCCCCAGGCCACGCCGTCTGCGGCAGCGCTGGAAGGAGGCGGTGGAGGAGGCGGCCAACGGCTCCCTGCCCCAGAAGGCCGAGTTCACCG
>CLocus_10056_Sample_934_Locus_12529_Allele_1 [sample_934; groupI, 49712]
TGCAGGCCCCAGGCCACGCCGTCTGCGGCAGCGTTGGAAGGAGGCGGTGGAGGAGGCGGCCAACGGCTCCCTGCCCCAGAAGGCCGAGTTCACCG
>CLocus_10056_Sample_935_Locus_13271_Allele_0 [sample_935; groupI, 49712]
TGCAGGCCCCAGGCCACGCCGTCTGCGGCAGCGCTGGAAGGAGGCGGTGGAGGAGGCGGCCAACGGCTCCCTGCCCCAGAAGGCCGAGTTCACCG
其中,红色标记处表示catalog locus,或这个locus在群体水平上的ID;绿色标记处表示sample ID,指出这个locus来自哪个样本,这个ID在Stacks流程中用于指示跟综样本,作为样本的编号;蓝色标记处表示样本内的locus ID;紫色标记处记录allele或者haplotype编号,样本名记录在中括号中。
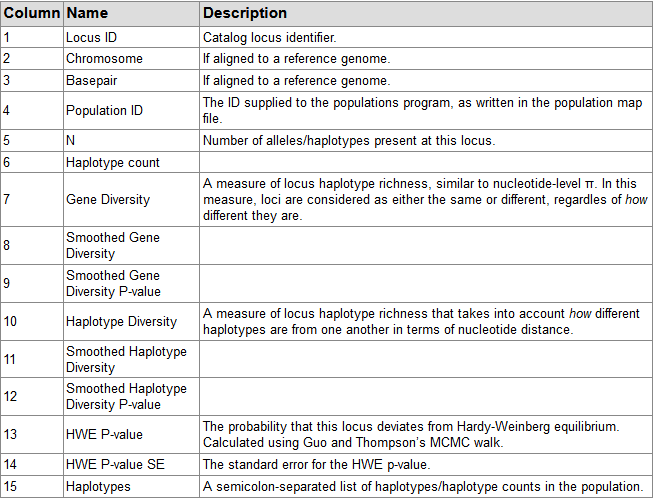
populations.hapstats.tsv: Haplotype-based summary statistics for each locus in each population
实例
某样本有22,338,004对PE reads,通过ustcks得到915,533条consensus序列,总的使用到的R1 reads有18,772,696;跟其它样本一起用cstacks处理后组装为3,766,982条consensus序列;再将该样本R1 match到catalog上,并融合PE reads最终得到3,674,170条融合后的tags。
参考材料:
[1] Nicolas C Rochette, Julian M Catchen. Deriving genotypes from RAD-seq short-read data using Stacks. Nature protocols. 2017, 12(12). 2640-2659
[2] J. Paris, J. Stevens, J. Catchen. Lost in parameter space: a road map for Stacks. Methods in Ecology and Evolution. 2017.
[3] https://github.com/lyl8086/RADscripts/blob/master/RADassembler/Tutorial.md
[4] Lost in parameter space: a road map for Stacks. Methods in Ecology and Evolution 2017.
[5] RAD sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Molecular Ecology Resources.
[6] http://catchenlab.life.illinois.edu/stacks/manual/
https://blog.sciencenet.cn/blog-2970729-1126523.html
上一篇:使用Biopython解析Blast结果
下一篇:HiCUP的使用以及结果解读