博文
2020年(5): 基于能量和结构的二维增强采样方法ossPTMetaD的发展与应用
||
蛋白质的功能发挥与其构象变化密切相关。因此,研究靶标蛋白的动态行为对理解其生物学功能以及药物发现与设计至关重要。分子动力学(MD)模拟能够从原子水平上监测蛋白质结构的动态变化,然而常规MD受限于其低采样效率,且易陷入局部极小值。增强采样方法通过各种原理提高MD采样效率,已成为研究蛋白质大规模构象变化的主流技术之一。其中,副本交换分子动力学(REMD)是一种通过多个平行模拟副本交互信息的增强采样方法,广泛应用于有机分子和生物大分子的构象采样及自由能的计算研究。但是REMD所需模拟副本数目与体系总自由度的平方根成正比,普通计算平台难以承受由此带来的巨大计算成本。
针对上述挑战,中科院上海药物研究所朱维良课题组提出了在能量和结构二维空间内增强采样的ossPTMetaD方法【1】,并将ossPTMetaD方法成功应用于3种不同运动类型的蛋白质模型体系,包括极具挑战性的激酶DFG翻转运动的模拟研究,所需计算资源约为传统REMD方法的30%。相关成果在12月22日在线发表于J. Chem. Theory Comput.杂志,论文第一作者为博士研究生彭诚。
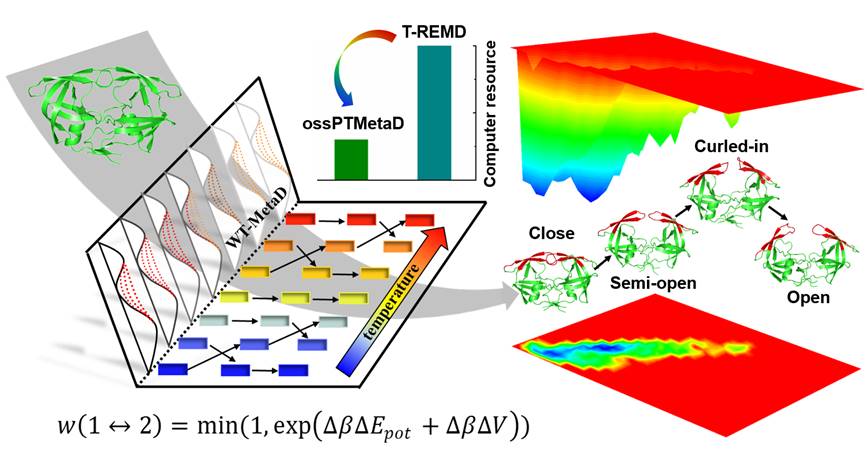
图1. ossPTMetaD方法示意图
溶剂分子在生物大分子模拟体系总自由度中占比往往远超60%,因此减少溶剂自由度是REMD优化的一个重要方向。为解决溶剂自由度问题,朱维良课题组前期发展了两种REMD优化方法,一种是基于速度调节的显隐式溶剂模型REMD方法【2】(以显式溶剂模型模拟体系运动,而以隐式溶剂模型计算副本交换概率,并通过速度调节关联这两种溶剂模型获得的能量差异),另一种是为了克服隐式溶剂模型缺陷而发展的vsREMD (Velocity-scaling optimized hREMD)方法【3】。在前期研究中,他们也发现简振模式分析(NMA)能够高效预测蛋白质的运动趋势,具有成为增强采样变量的潜力。因此,本工作通过整合REMD与另一种高效的增强采样方法(Well-tempered Metadynamics),并且采用NMA作为增强采样变量,发展了ossPTMetaD方法(图1)。新方法在副本交换时也去除溶剂内部作用能的影响,显著减少了体系的自由度及所需的副本数目。
采用ossPTMetaD方法,作者首先研究了腺苷酸激酶(AdK)关闭(closed)与打开(open)两种构象的动态转换过程。相比REMD方法需要80个模拟副本,ossPTMetaD只需24个副本即可成功模拟此构象变化(图2),并且具有比REMD更高的副本交换率和更快的模拟收敛性。作者将腺苷酸激酶不同状态的实验结构映射入自由能图景中发现,这些实验结构均在ossPTMetaD模拟预测的构象转变路径附近,进一步表明ossPTMetaD模拟结果的合理性。
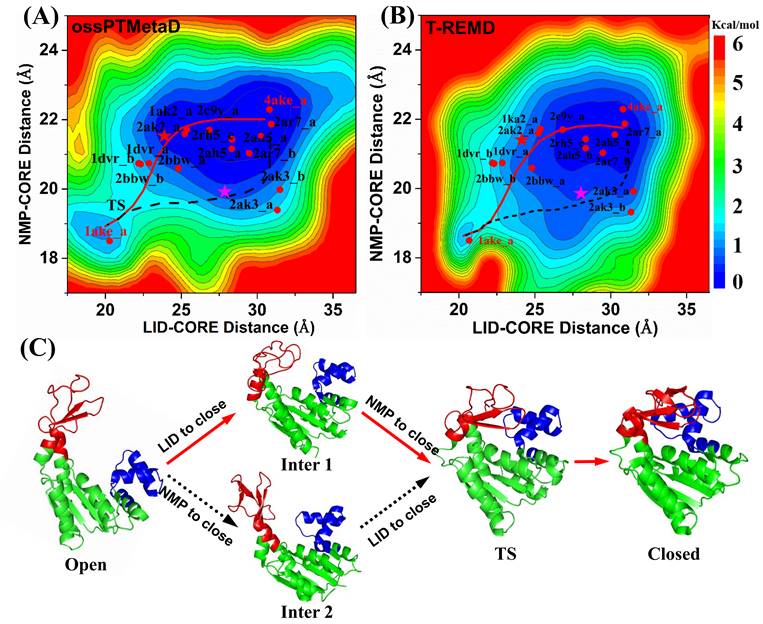
图2. 腺苷酸激酶构象变化的模拟结果。A和B分别是ossPTMetaD方法与传统REMD方法获得的腺苷酸激酶构象变化自由能图景。C是腺苷酸激酶从开放构象转变成闭合构象的两条主要低能路径。
作者进一步将ossPTMetaD方法用于模拟HIV-1蛋白酶和BACE1蛋白的flap柔性运动以及激酶的DFG翻转运动。对于HIV-1蛋白酶,作者通过ossPTMetaD方法获得了4种不同的HIV-1蛋白酶低能构象以及它们之间的相互转变能垒。其中,除了目前实验已经观察到的闭合构象和半打开(semiopen)构象外,作者还发现了另外两种低能构象:curled-in 和fully open构象。BACE1蛋白被认为是一种治疗阿尔茨海默症的重要靶点,作者通过ossPTMetaD方法不仅研究了BACE1空蛋白的flap柔性变化,并且发现不同结构的BACE1抑制剂能够将蛋白结构限制在闭合和打开两种不同的状态。对于激酶体系,作者通过ossPTMetaD方法成功模拟了p38α和c-Abl两种激酶的DFG翻转运动,并获得这类构象变化的自由能图景以及中间低能构象。同时本文作者也指出,不同于传统REMD,ossPTMetaD在副本交换时并未严格遵从细致平衡条件(Detailed Balance Condition),因此需要更多的体系测试以进一步验证该方法的准确性和普适性。
全文链接:https://doi.org/10.1021/acs.jctc.0c00592
参考文献
1. Peng, C.; Wang, J.; Shi, Y.; Xu, Z.; Zhu, W. Increasing the Sampling Efficiency of Protein Conformational Change by Combining a Modified Replica Exchange Molecular Dynamics and Normal Mode Analysis. J. Chem. Theory Comput. 2020, https://doi.org/10.1021/acs.jctc.0c00592
2. Wang, J.; Zhu, W.; Li, G.; Hansmann. U.H. Velocity-scaling optimized replica exchange molecular dynamics of proteins in a hybrid explicit/implicit solvent. J. Chem. Phys. 2011,135, 084115.
3. Wang, J.; Peng, C.; Yu, Y.; Chen, Z.; Xu, Z.; Cai, T.; Shao, Q.; Shi, J.; Zhu, W. Exploring Conformational Change of Adenylate Kinase by Replica Exchange Molecular Dynamic Simulation. Biophys. J. 2020, 118, 1009.
https://blog.sciencenet.cn/blog-2877557-1265641.html
上一篇:2020年(4): 三氟甲基取代对化合物活性的影响
下一篇:JCIM 60周年纪念刊