博文
2012ISME:利用网络分析探讨土壤微生物群落共现模式
||
Using network analysis to explore co-occurrence patterns in soil microbial communities
利用网络分析探讨土壤微生物群落共现模式

摘要
探索由高通量DNA测序技术产生的大型环境数据集需要新的分析方法,以超越对自然微生物群落组成和多样性的基本清单描述。为了研究微生物类群之间潜在的相互作用,对重要类群共现模式的网络分析可能有助于破译跨空间或时间梯度的复杂微生物群落结构。在这里,我们计算了微生物类群的相关性和对一个16S rRNA基因条形码焦磷酸测序数据集应用了网络分析方法,该数据集包含来自广泛生态系统类型151个土壤样本的4160000个细菌和古细菌序列。我们描述了由此产生的网络的拓扑结构,并基于丰度和占有率(即生境普遍物种和生境特有物种)定义了操作分类单位类别。共现模式很容易被揭示,包括一般的非随机关联、广泛的分类水平上的共同生活史策略和社区成员之间的意外关系。总的来说,我们展示了探索类群间相关性的潜力,以便更全面地了解微生物群落结构和指导群落组装的生态规则。
前言
近年来,对复杂微生物群落的研究取得了长足的进展,部分原因是方法学的进步,例如高通量DNA测序技术产生了有关微生物群落组成的详细信息(Sogin等人,2006年)。序列数据通常来源于对一部分小亚单位rRNA基因进行测序(Pace,1997),并且可以应用多种技术对序列数据进行分析,以描述微生物群落的组成、多样性以及群落在空间、时间、实验处理上的变化。然而,大多数分析技术都集中在研究群落的单一特性上。例如,描述和比较微生物群落结构的研究通常侧重于单个样本中发现的分类群或独特谱系的总数(即α多样性),个体分类单元或谱系的相对丰度以及群落或群落类别之间的系统发育或分类重叠程度(即β多样性)。α多样性测度(例如丰富度和覆盖度估计器、稀疏曲线)对不同环境中微生物多样性及其限度的产量估计(Hughes等人,2001年;Curtis等人,2002年;Sogin等人,2006年)。同样,多元统计技术,如聚类和排序,使微生物生态学家能够描述β多样性模式,揭示生物和非生物变量如何控制微生物群落组成。例如,对β多样性模式的分析揭示了微生物群落是如何在广泛的自然栖息地中构建的(Lozupone and Knight,2007;Auguet et al.,2010;Barbera'n and Casamayor,2010,2011),人体内外微生物群落的时空变异性(Fier et al.,2008;Costello等人,2009),以及构成土壤细菌群落的因素(Lauber等人,2009)。相反,人们很少关注使用序列数据来探索环境样品中共存的微生物类群之间的直接或间接相互作用。记录复杂多样群落中类群之间的相互作用(即共现模式)可能有助于确定未培养微生物所占据的功能角色或环境生态位(Ruan等人,2006年;Fuhrman和Steele,2008年;Chaffron等人,2010年)。随着微生物群落序列数据的不断积累,我们现在面临着开始探索这些相互作用的挑战,并将群落分析扩展到探索代表大多数基于序列的微生物群落分析的α和β多样性模式之外。
网络分析工具和网络思维已被生物学家、数学家、社会科学家和计算机科学家广泛用于探索实体之间的相互作用,这些实体包括学校中的个体,食物网中的物种,计算机网络上的节点,或代谢途径中的蛋白质。网络分析用于探索一组项目(节点)的数学、统计和结构特性以及它们之间的联系。除了几个显著的例外,网络分析尚未广泛应用于探索复杂群落中微生物类群之间的共生模式。为了通过网络分析检测栖息地内和栖息地之间微生物之间的牢固联系,有必要对相对大量样品中发现的微生物类群有相当详细的信息,因为没有足够大的样本集,很难确定共现模式是否具有统计学意义。理想情况下,样本集应覆盖环境条件下的空间或时间梯度,以便在分类单元丰度上有足够的变异性来解决共现模式。最近使用条形码焦磷酸测序法来调查大量样品中微生物群落的研究表明,现在有可能生成能够充分利用网络分析方法的微生物数据集,我们甚至可以将它们应用于高度多样化的群落,如土壤中发现的群落,以探索共生模式。分类群共现模式的网络分析提供了对复杂微生物群落结构的新见解,补充和扩展了标准分析方法提供的信息。首先,分类群间的联系可能有助于揭示社区成员(甚至是不同生活领域的成员,如细菌和古细菌)共享的生态位空间,或者,也许是社区成员之间更直接的共生关系。在土壤等环境中,这些信息尤其有价值,因为许多微生物类群的基本生态和生命史策略仍然未知。探索土壤微生物之间的共生模式有助于识别潜在的生物相互作用、栖息地亲缘关系或共享的生理学,从而指导更集中的研究或实验环境。更普遍地说,网络分析代表了一种在大型复杂数据集中探索和识别模式的方法,使用广泛应用于微生物生态学的标准α/β多样性指标可能更难发现模式。在这里,我们使用网络分析来探索土壤中的原核分类群之间的联系,土壤是地球上最复杂和富含分类群的微生物栖息地之一。我们分析了来自不同生态系统类型151个土壤样本的16万多个细菌和古细菌16S rRNA基因序列,以证明网络分析的实用性,并解决以下问题:(i)土壤微生物的共生是否比预期的更偶然?(ii)观察到的和随机的种内共现之间缺乏一致性是否可以作为生态位分化的一种替代?和(iii)哪些分类群是普遍物种(广泛分布于土壤生境)或特有物种(局限于某些生境但局部丰富)以及这些生态类别如何形成网络结构?
材料和方法
土壤描述和分子方法
该数据集包括分布在南北美洲和南极洲的151个土壤样本。收集的土壤来自广泛的生态系统、气候和土壤类型(补充表1)。土壤收集方法和土壤和环境特性的方法已经在前面描述过(Fier和Jackson,2006;Bates等人。,2010年)。提取DNA的制备遵循Fiereret al.,2008和Bates et al.,2010中详细描述的方案。简言之,16S rRNA基因的一个区域用引物F515(5'-GTGCCAGCMGCCGCGGTAA-3')和R806(5'-GGACTACVSGGGTATCTAAT-3')扩增,该引物应能扩增几乎所有细菌和大肠杆菌,且对特定群体几乎没有偏见(Bateset al.,2010)。得到的条形码PCR产物以等摩尔量标准化,并在南卡罗来纳大学环境基因组学技术平台(Engencore)的Roche GS-FLX 454自动焦磷酸测测序仪(Rochepplied Science,Branford,CT,USA)上进行测序。
序列处理
在QIIME中处理了从焦磷酸测序产生的原始序列数据(Caporaso等人,2010年)。
简单地说,使用uclust的90%阈值,对序列进行质量修剪并聚类成可操作的分类单元(OTUs)(Edgar,2010)。90%的识别阈值与细菌科的分类水平大致对应(Konstantindis和Tiedje,2007),用于生成一致的高丰度OTU,用于基于相关性的后续分析,并避免测序异常导致的潜在分类错误。如果我们使用更标准的‘物种水平’OTU阈值(97%的序列一致性),得到的OTU表会更大,使得数据可视化和分析更加困难。在90%的同一性水平上,最终的OTU表包含160469个序列(每个样本平均1063个序列),分布在4088OTU中,其中2798个序列由1个以上的序列表示。分类分配使用RDP分类器进行(Wang等人,2007年),并通过对GenBank非冗余核苷酸数据库(nt)的BLAST搜索进行手动管理。
数据分析
非随机共现模式用棋盘评分(C-score)在保持位点频率的零模型下进行测试(Stone andRoberts,1990)。一个棋盘单元是一个2×2矩阵,其中两个OTUs发生一次,但在不同的站点。对于网络推断,我们计算了超过5个序列的OTU(1577个OTU)之间所有可能的Spearman秩相关。之前的过滤步骤去除了不良的代表性,降低了网络复杂性,有助于确定核心土壤群落。如果Spearman的相关系数(r)>0.6且统计意义重大(P<0.01;Junker和Schreiber,2008),我们认为共现事件是有效的。重建网络中的节点代表90%相似的OTUs,而边(即连接)对应于节点之间强大而显著的相关性(有关最终的网络,请参阅GRAPHML格式的补充文件)。为了描述产生的网络的拓扑结构,计算了一组度量(即平均节点连接性、平均路径长度、直径、累积度分布、聚集系数和模块性)(Newman,2003)。所有的统计分析都是在R环境下使用vegan和igraph包进行的。使用交互式平台gephi对网络进行了探索和可视化(Bastian等人,2009年)。
结果和讨论
一般共现模式
土壤是一种异质环境,蕴藏着极其多样的原核生物群落(Torsvik等人,1990年;Curtis等人,2002年)。以前的工作从不同的角度探索了土壤微生物多样性,包括估算单个样品中的物种丰富度水平(例如,Fier等人,2007年;Roesch等人,2007年;Youssef和Elshahed,2009年),评估控制群落多样性和组成的非生物变量(例如,McCaig等人,2001年;Fier和Jackson,2006年;Lauber等人,2009年),以及特定非生物因素如何影响特定分类群的评估(例如,Jones等人,2009年)。微生物类群之间的关系也塑造了微生物群落的结构(Prosser等人,2007),因此,可以预期,非随机共生模式和显著的类群间关系应该出现。在以90%的一致性进行质量过滤和OTU聚类之后,我们获得了2798个OTU,由分布在本研究所包括的151个土壤样本中的一个以上序列表示(补充表1)。为了评估非随机共现模式,我们首先使用了基于棋盘格单位的生态测量(C分数;Stone和Roberts,1990)。总的来说,我们使用整个数据集观察到非随机共现模式(C评分为1/4 46.56,P值为0.01)。将分析局限于那些显示出显著关系的OTU(如图1所示),测量值增加到C评分为1/4 185.03,P值为0.01。最近的一项宏分析显示,微生物和大型生物的共生模式相似,表明非随机群落聚集可能是所有生命域的一个普遍特征(Horner Devine等人,2007)。这一发现表明存在显著的非随机共现模式并不奇怪,因为我们已经知道了一段时间,许多细菌类群显示出可预测的生物地理模式(Prosser等人,2007)。同样,记录非随机共现模式与实际确定构成社区的因果机制有很大不同。然而,非随机装配模式确实表明了确定性过程的优势,包括竞争性相互作用、不重叠的生态位或形成群落组成的历史效应(Horner Devine等人,2007)。总的来说,这种方法使我们能够得出这样的结论:土壤微生物的共现往往比预期的多。
网络描述
一旦我们确定土壤微生物组合模式肯定是非随机的,我们就进一步利用基于强相关性和显著相关性的网络推断探索共生模式(使用非参数Spearman's;Junker和Schreiber,2008)。共生微生物的相关网络允许对大量信息进行可视化摘要(Chaffron等人,2010),并已成功应用于识别海洋微生物与其环境之间的关联(Ruan等人,2006)。
由此产生的土壤微生物网络(图1和补充图1)由296个节点(OTU)和679个边缘组成(平均度或节点连接性4.59;累积度分布见补充图1)。计算了网络分析中常用的一些拓扑性质来描述OTU之间复杂的相互关系模式(Newman,2003)。所有节点对之间的平均网络距离(平均路径长度)为5.53条边,直径(最长距离)为18条边。聚类系数(即节点如何嵌入到其邻域中,因此它们倾向于聚集在一起的程度)为0.33,模块化指数为0.77(值>0.4表明网络具有模块化结构;Newman,2006)。总体而言,土壤微生物网络由高度连接的OTU(每个节点~5条边)组成,OTU结构在密集连接的节点组(即模块)之间,并形成一个集群拓扑结构(正如对比随机图更显著集群化的现实网络的预期)。这些结构特性为不同生态系统类型的复杂数据集之间的快速和简单的比较提供了潜力,以便探索某一生境类型的一般特征如何影响微生物群落的聚集。
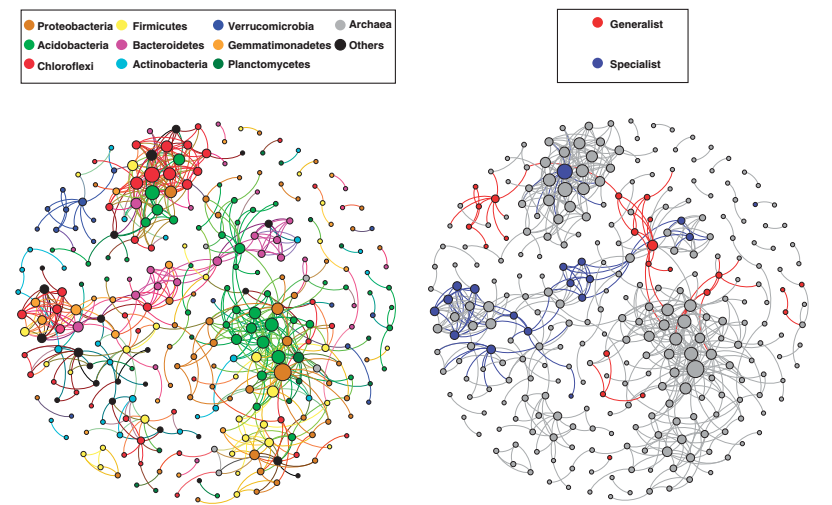
图1基于相关分析的共现90%阈值OTU的网络。一个连接代表一个强大的(斯皮尔曼r>0.6)和显著(P<0.01)的相关性。每个节点的大小与连接数(即度)成比例。左图:按分类着色的OTUs。右图:按丰度和占有率着色的OTU(普遍物种和特有物种)。
结构分析还表明,当考虑到观察到的门频率和随机关联时,来自同一个门的OTU往往比预期的要多(33%)(11%;图1,左面板)。因此,一方面,观察到的显著共现的门内百分比与随机关联下的预期聚集之间缺乏一致性的程度,另一方面,可以用作生态位偏好或协同关系的指标。该指数还可以反映不同生境(例如,水生与土壤)之间的差异,从而阐明指导微生物群落组成的生态规则。在所有的可能性中,这些共现模式中的大多数来自于共享相似生态位的分类群,而不是直接共生体,然而,仅仅这些数据不允许我们将这两种可能性分开。一些共生模式揭示或证实了尚未被很好研究的分类群的有趣生态模式。例如,疣状支原体门的成员往往比偶然出现的(0.3%)多(1.8%),这表明,尽管它们在土壤中丰富且普遍存在(Bergmann等人,2011年),但它们共享一个特定的(尚未定义的)生态位(图4)。另一个丰富的世界性的门,也显示出比随机关联的预期更高的共生发生率是酸杆菌(2.4%随机,9.4%观察到;图4)。在这种情况下,这种模式很可能是由之前观察到的现象驱动的,即土壤pH值在很大程度上控制着许多土壤酸杆菌分类群的分布(Jones等人,2009)。其他例子是氯磷菌和球菌群,既不是丰富的,也不是普遍物种/世界主义(图4),而是,似乎主要限于沙漠土壤。几个OTU共享相同的栖息地偏好,因此似乎是非常互联的(1.9%随机,而6.2%的氯氟克利;0.01%随机,而1%观察到球菌)。因此,观察到的和随机共现之间的不一致程度可以为在不同关联水平上共享同一系统发育的不同种群的生态位分化提供进一步的见解。总的来说,这些发现表明,环境过滤效应和生态位分化在广泛的分类水平上是明显的,正如其他地方所指出的(Philippot等人,2010)。
栖息地普遍物种和特有物种
在丰度与占有率图(图2)中,每个由多个序列代表的OTU被划分为两大类:,一个是土壤通才(即广泛分布的微生物类群,我们在操作上将其定义为在151种土壤中的>80种土壤出现),另一个是土壤专家(在操作上定义为当地丰富的土壤,代表单个库中>2%的序列,但仅在<10个土壤样本中发现)。根据这一标准,~2%的OTU属于通才类别,~1%属于专科类别。尽管在本研究中使用了一种比传统方法具有更高测序深度的高通量DNA测序方法,但我们可能遗漏了可能是普遍物种的极低丰度分类群。然而,将生态群落中的分类单元划分为这两个类别(我们承认这两个类别的定义有些武断)对于定义生态类别/策略是有用的,这些类别/策略提供了系统发育、分类或功能能力定义的信息(Magurran和Henderson,2003)。最近,基于丰度和占有率的微生物分类在临床样本分析中被证明是有用的(van der Gast等人,2011)。在一般生态学中,在许多空间尺度上观察到平均丰度和占有率之间的正相关关系(Guo等人,2000;van der Gast等人,2011)。然而,我们没有在我们的数据集中观察到这种趋势。虽然本研究所分析的土壤样本大多来源于温带和肥沃的土壤,但本研究所涵盖的环境变异(即不同的生境和广阔的空间尺度)可能改变了这种关系。例如,居住在沙漠或南极洲土壤等极端环境中的专业细菌OTU的丰度比预期的要高在整个采样范围内的持久性。
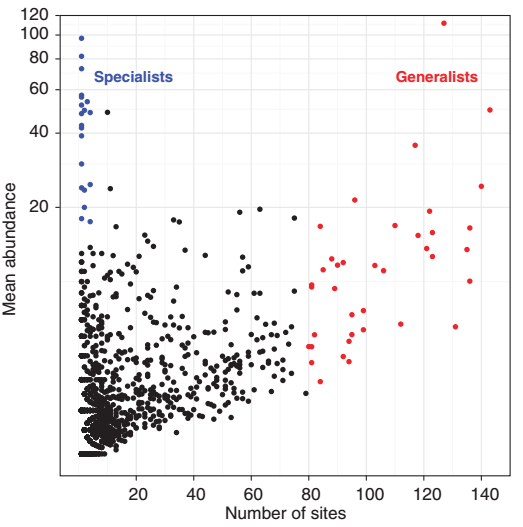
图2 90% OTU的丰度(y轴)和占有率(x轴)图。普遍物种OTUs(红色)定义为出现在480个土壤样本中。特有物种OTUs(蓝色)定义为当地丰富(418个序列)和出现在10种土壤中。
总的来说,我们观察到普遍物种和特有物种在共现网络分析中的一致性分离(参见图1右面板中与所有剩余OTU的显著相关性,以及图3中添加分类信息的细节)。特有物种OTUS(本地丰富的几个样品)是由不同的系统发育组组成的不常见的土壤中的大多数生物群落(即,Chloroflexi, Deinococcus, Gemmatimonadetes;图3)。普遍OTUs(广泛分布)依次是来自酸杆菌、变形菌(尤其是α亚类)和疣微菌菌群的典型土壤成员(参见图4,顶部和底部面板,以及Janssen,2006年的最新评论)。不同的分类组成和分布范围可能影响观察到的网络结构(图3),表明这两个类别对网络结构的影响不同。
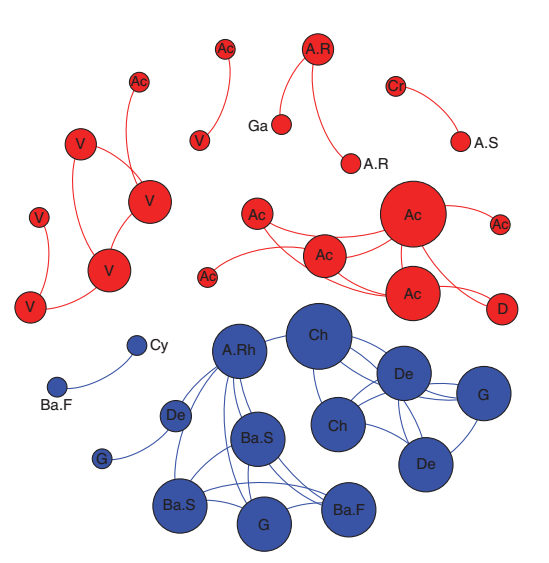
图3基于相关分析的普遍物种和特有物种90% OTU的共现网络。一个连接代表一个强大的(斯皮尔曼的r>0.6)和显著(P<0.01)的相关性。每个节点的大小与连接数(即度)成正比。根据分类学的标签:Ac,酸杆菌门。A.R,变形菌门,根瘤菌目。A.Rh,α变形菌门;红杆菌目。A.S,α变形菌门;鞘氨醇单胞菌。Ba.F,拟杆菌;黄杆菌。Ba.S,拟杆菌;鞘氨醇杆菌。Ch,绿弯菌门。Cr,泉古菌门。Cy,蓝藻;D,δ变形菌门。De,异常球菌属。G,芽单胞菌门。Ga,γ变形菌门。V,疣微菌门。
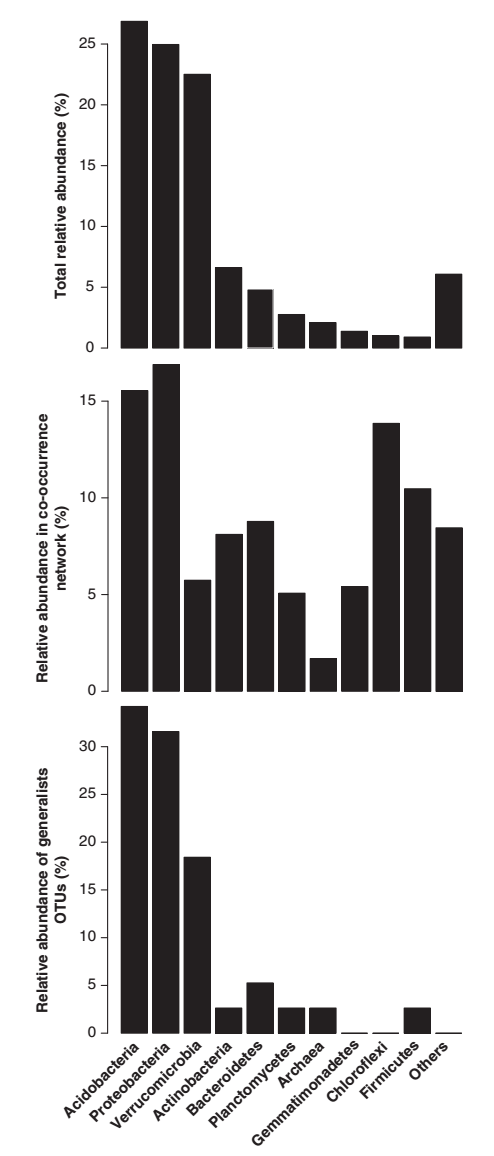
图4不同微生物分类群的相对丰度。顶面板:所有土壤中的序列数样品。中间面板:显著同时出现的OTU数量(图2中的节点)。低面板:普遍性OTU的数量。
与特有物种网络(29个显著共现和2个分区)相比,普遍物种网络的连接较少,且更为划分(19个显著共现和5个分区),这可能是因为前者覆盖的生境变异性最高,而后者在受限环境中存在。因此,我们所建立的两个任意的生态分类允许我们捕捉到关于群落聚集结构的额外信息,如之前所示的大型生物(Pandit等人,2009)。最后,通才OTU与整个数据集之间的显著相关性如表1所示。列出的相关性不包括同一分类组成员之间的共现事件。普遍物种OTUs属于疣状芽孢杆菌、酸杆菌、变形菌和拟杆菌等丰富的门。有趣的是,没有被归类为典型和丰富土壤成员的普遍物种OTUs(如Deltaproteobacteria和Crenarchaeota)与其他无处不在的成员共同出现。crenarchaeotal OTU(与Candidatus Nitrososphaera gargensis密切相关,以前被描述为在土壤中普遍存在;Bates等人,2010年)的情况尤其显著,因为我们对该分类群所占的生态位了解不足,即使已经提出,相关的克氏古菌可能作为氮氧化剂在氮循环中具有重要作用(莱宁格等人,2006)。与甲烷氧化密切相关的序列共发生了这一特殊现象(表1)。这一观察可能是第一个步骤,在土壤硝化过程中修正土壤Cern古生菌的预期功能作用,因为催化氨氧化(氨单加氧酶)和甲烷氧化(颗粒甲烷单加氧酶)的酶的高序列相似性;福尔摩斯等人,1995)。这是一个潜在的例子,即本研究中提出的方法必须获得关于难以捉摸但与生态有关的微生物的知识。
表1普遍性OTU分类(图3中红色节点)及其显著性(P值<0.01)共现OTU

最后的评论
通过这项工作,我们证明了将网络分析方法纳入到目前为止微生物生态学家可用的统计方法库中的效用。通过对一个由焦磷酸测序产生的大型土壤微生物数据集进行网络分析,探索复杂数据集的过程更加可行,并且出现了有趣的未知模式,包括非随机关联、不同组分水平的确定性过程以及社区成员之间的意外关系。指导微生物群落组成的不同生态规则可能反映在不同的生境网络结构中(例如,水生与土壤,或早期殖民与后期演替生态系统),值得进一步研究。下一个合乎逻辑的步骤是超越仅仅描述通过网络分析和设计更具针对性的实验所揭示的模式,或者研究特定的环境梯度和社区随时间的变化,以便理解产生社区共存模式的机制,也就是说,究竟是什么决定了一个群落中哪些物种和多少物种共同生活。
https://blog.sciencenet.cn/blog-2675068-1243091.html
上一篇:带有显著性标记的相关性热图绘制方法
下一篇:[转载]解决 Endnote 的 Word 插件 不自动加载