博文
如何使用Bioconductor进行单细胞分析?
|

最近的技术进步使得能够在单个细胞中分析全基因组特征。但是,单细胞数据为分析提出了独特的挑战,需要开发专用的方法和数据架构才能成功解析数据背后的生物问题。Bioconductor项目托管了社区开发的开源R包以满足这些分析需求。我们为潜在用户提供了单细胞方法的概述和在线图书,内容涵盖最先进的计算方法、标准化的数据基础架构和交互式数据可视化工具,存储于 https://osca. http://bioconductor.org。
从2001年开始,Bioconductor项目已经吸引了来自不同科学领域的众多开发人员和用户社群,推动了使用R语言进行高通量生物数据分析的开源软件包的开发。尽管大量组学技术催生了重要的科学见解和研究方法,近来单细胞组学图谱的发展可以回答以前无法回答的科学问题。Bioconductor拥有大量用于分析组学数据的软件包,最近,随着社区贡献的软件包迅速增多,Bioconductor已显著扩展到单细胞数据分析领域(图1)。
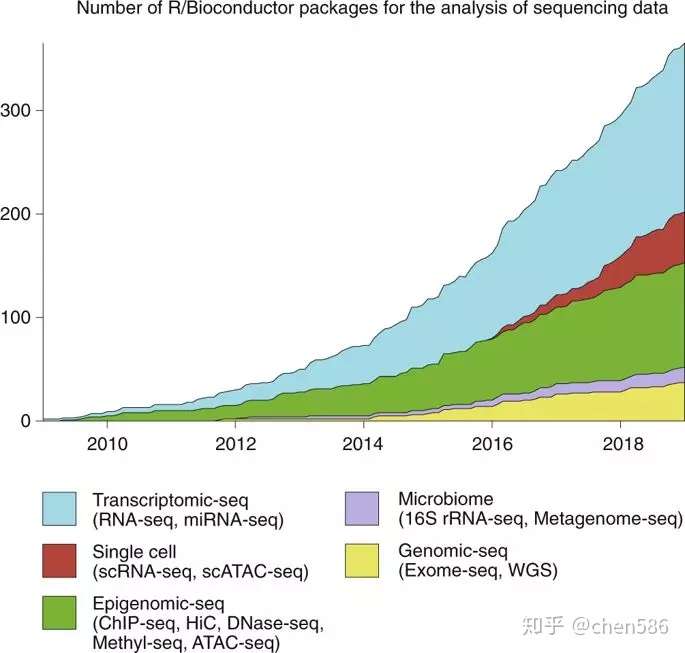
Fig. 1 | Number of Bioconductor packages for the analysis of highthroughput sequencing data over ten years.
当前的单细胞测序既可以是高通量的,同时测量数千到数百万个细胞;也可以是高维的,同时测量单个细胞内的数千个特征。与大量细胞样品的图谱相比,单细胞数据有两个特征,必须对其进行特殊处理才能获得有意义的生物结果:(1)数据中的细胞数目增加了多个量级,如人类细胞图集和小鼠细胞图集等;(2)由于所测量特征(基因/转录本)的生物波动性高或对少量分子进行定量分析的敏感性有限,导致数据稀疏性增加。这些特性促进了针对单细胞数据分析的统计方法的发展。此外,随着单细胞技术的成熟,数据复杂性和数据量的增加要求对数据访问、管理和基础架构进行根本性的改变,同时需要专门的方法来促进可扩展的分析。
为了应对这些挑战,为分析单细胞数据而开发的软件包已成为Bioconductor项目不可或缺的一部分。在这里,我们主要集中在分析单细胞RNA测序(scRNA-seq)数据,其中提到的许多概念也可推广到其他类型的单细胞项目。我们的描述涵盖了数据导入、存储单细胞实验数据的通用数据结构和用于将原始单细胞数据转换为适合下游分析、交互式数据可视化和下游分析的快速而强大的方法。为了帮助用户利用这个强大且可扩展的框架,我们介绍了选定的软件包并提供了在线图书(https://osca.bioconductor.org),内容涵盖软件包安装、使用帮助、特定scRNA-seq分析主题和分析各种scRNA-seq数据集的完整工作流程。有关所有软件包的参考资料,请参见:http://bioconductor.org/packages/.
数据结构
Bioconductor的一个强大的优势是提供了一个复杂的、高度互相依赖的数据集的通用表示形式和基础架构。Bioconductor使用标准化的数据容器来实现各种包的模块化和交互操作,同时保持强大的终端用户易用性。为此,Bioconductor采用了一种称为S4的灵活的面向对象范式,可以使用丰富且用户友好的方式将多个对象组件封装到单个实例中。这种方法对于生物学分析尤其重要,因为在整个分析过程中,数值数据和元数据需要在整个分析过程中都维持一致性。
Bioconductor使用SingleCellExperiment类来存储单细胞测序数据和元数据(图2). 诸如计数矩阵之类的主要数据以一个或多个矩阵的形式存储在assay组件中,其中行代表特征(例如基因和转录本),列代表细胞。此外,基本数据的低维形式和描述细胞或特征属性的元数据也可以存储在SingleCellExperiment对象中。通过SingleCellExperiment类,可以将与scRNA-seq实验相关的所有数据和结果存储在单个实例中。通过单细胞数据和结果的标准化存储,Bioconductor促进了单细胞分析程序包之间的交互性,并促进了复杂分析工作流程的开发和使用。
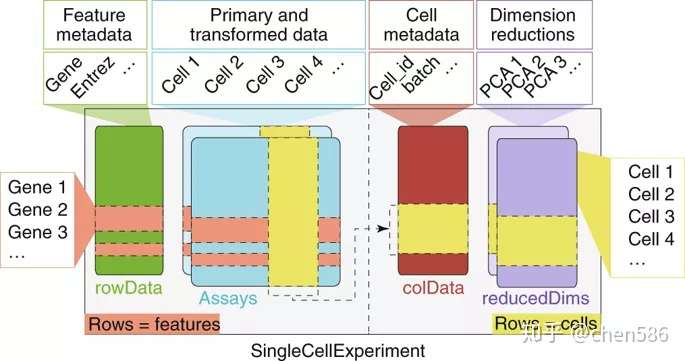
Fig. 2 | Overview of the SingleCellExperiment class.
数据处理
本节的目的是描述大多数scRNA-seq分析所共有的前期分析步骤。这些基本步骤遵循通用的分析流程(图3):(1)预处理原始测序数据生成每个基因(或转录本)X 每个细胞的表达计数矩阵,然后创建SingleCellExperiment对象;(2)对数据进行质控并去除可能会干扰下游分析的低质量细胞;(3)将原始计数转换为标准化的表达值,以消除细胞和基因特异性偏好;(4)进行特征选择筛选生物学相关基因进行下游分析;(5)应用降维方法压缩数据并降噪;(6)如果需要,整合多批次scRNA-seq数据。
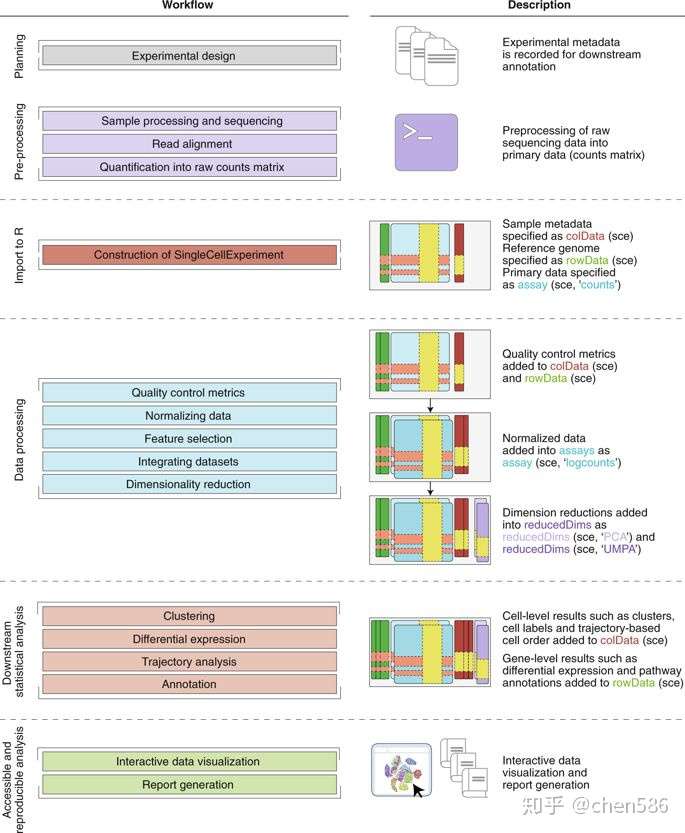
Fig. 3 | Bioconductor workflow for analyzing single-cell data. A typical analytical workflow using Bioconductor leads to the creation and evolution of a SingleCellExperiment (sce) object during data processing and downstream statistical analysis (left column). An example of an sce object evolving throughout the course of a workflow is shown, including visualization, analysis and annotation (right column).
预处理。对于scRNA-seq数据,预处理包括将测序reads与参考转录组进行比对,然后获得每个细胞和每个基因的表达值计数矩阵。尽管多种命令行软件形式的预处理方法已经存在,scPipe和scruff等Bioconductor软件包提供了完全用R编写的预处理工作流。DropletUtils和tximeta等Bioconductor软件包可以读入各种命令行软件工具如Cell Ranger) (10X基因组学),Kallisto-Bustools和Alevin的结果。值得注意的是,伪对齐(pseudo-alignment )方法(例如Alevin和Kallisto)显著减少了计算时间和运行内存。
在上述所有工作流程中,最终结果是将计数矩阵导入R并创建SingleCellExperiment对象。对于特定的文件格式,我们可以使用DropletUtils(用于10X数据)或tximeta(用于伪对齐方法)包中的专用方法。
质量控制。造成scRNA-seq数据中的低质量文库可能有多种原因,如解离时的细胞损伤或文库制备失败(例如,不成功的逆转录或PCR扩增)。这些通常表现为“细胞”的总计数低、表达的基因数目很少、线粒体基因表达占比高。这些低质量的文库可能会导致下游分析中获得误导性结果。
对于基于液滴的实验方式,通常只保留包含且只包含一个细胞的液滴生成的数据。DropletUtils程序包根据观察到的每个液滴的表达谱与周围溶液的表达谱来区分空的(只含溶液中RNA的)液滴和含细胞的液滴。它还可以去除基于液滴的实验中由于barcode序列错误产生的假细胞。同样,scran或scds程序包可以比较实验检测到的液滴与模拟的doublets液滴的表达谱识别可能包含多个细胞(doublets)的液滴。在排除空液滴并识别潜在的doublets后,将含有潜在受损细胞或测序覆盖度较差的液滴过滤掉。库大小(定义为每个细胞所有相关基因的总计数之和)是一个常用的过滤指标。具有较小文库大小的细胞更可能是低质量细胞,这可能是因为在RNA制备过程中的某个步骤如细胞裂解、不成功的cDNA捕获和扩增等造成了RNA丢失。另一个指标是每个细胞中表达的基因的数量,定义为该细胞中具有非零计数的内源基因的数量。表达基因很少的细胞可能是转录本群体没有被成功捕获。线粒体基因组中基因的表达比例也是一个指标,因为线粒体基因比例高可能是因为细胞损伤造成细胞质RNA丢失,而线粒体因为体积大于单个转录物分子不太可能通过细胞膜上的孔逸出。Scater软件包简化了这些指标的计算。
标准化。scRNA-seq数据不同文库之间存在覆盖率的系统差异,例如测序深度差异。这通常是由于细胞之间cDNA捕获或PCR扩增效率不同而引起的,而这又是由于起始RNA量低导致的。标准化的目的是消除这些系统差异,以使它们不干扰聚类或差异表达分析时细胞之间表达谱的比较。
我们先只考虑在单个scRNA-seq实验中降低系统差异的方法,因为它们造成数据的偏好性的原因相似。例如,测序深度的变化将所有基因的表达计数按一定因子进行缩放。文库大小归一化是最简单策略,如scater中所实现。尽管此方法假设任何一对细胞之间的差异表达基因(DEG)上下调平衡(基因整体表达量不变),但是标准化准确性通常不是scRNA-seq探索性分析的主要考虑因素,因为它们对簇聚类的影响很小。
但是,准确的标准化在解释每个基因的统计数据如差异基因分析时非常重要。当在一个给定的scRNA-seq数据集中存在多种细胞类型时,最经常观察到表达偏差是表达变化对数值的偏移。通过反卷积进行归一化可以克服这一点,方法是合并许多细胞中的计数数据增加计数的大小以进行准确的size factor估计,然后将其解卷积为基于细胞的因子以对每个细胞进行标准化(如在scran中实现).
另外,BASiCS, zinbwave和MAST提供了基于模型的标准化方法,不仅可以处理此类文库大小或组成偏差,还可以针对已知的协变量或其他可能干扰生物学上有意义的变异的技术因素进行校正。这些方法支持更复杂的标准化策略,例如数据的非线性转换。有关此主题的评论,请参考(42).
缺失数据填充 (imputation)。数据插补方可以用来解决单细胞测序数据的稀疏性问题。由于scRNA-seq实验经常无法测量到某些基因的表达,从而导致数据表中零值过多,为此开发了零膨胀模型(zero-inflated models)。但是,其效果取决于检测方法或protocol的类型,尚无适应所有数据的最优工具。此外,研究表明,scRNA-seq数据的插补方法会导致假阳性结果,并降低了细胞类型特异性标记基因鉴定的可重复性.
特征选择。scRNA-seq数据的探索性分析通常旨在表征细胞间的异质性。诸如聚类和降维之类的分析会根据细胞的基因表达谱进行比较。但是,在这些计算中选择哪些基因用于下游分析影响重大。特征选择方法旨在识别能对研究的生物系统提供有用信息的基因,同时删除导致随机噪声的基因。通过只对此类基因进行分析,可以在排除排除混淆信息的基础上保留有意义的生物学结构。此外,只关注转录组的这一子集可以显著减小数据集的大小,从而提高下游分析的计算效率。参见(50,51)有关特征选择方法的评论。
特征选择的最简单方法是根据基因在整个细胞群体中的表达来选择变化最大的基因。这基于一个假设,真正的生物学差异导致的基因表达变化大于其他仅受技术噪声影响或无关的生物因素引起的表达变化。但是,对数转换无法实现完美的方差稳定化(variance stabilization)。这意味着相比生物异质性,基因的丰度对其程度影响更大。因此,特征选择计算每个基因的方差时通常需要对均-方差关系进行建模。软件包scran,BASiCS 和scFeatureFilter都采用这种方法。
另外,还有可以替代方差的度量标准,例如基于基因的偏离度(deviance)选择特征基因,该方法评估每个基因与细胞间恒定表达的零模型(null model)的拟合程度。与基于方差的特征选择方法不同,偏离度的计算是根据原始的唯一分子标识符(UMI)计数完成的,因此该方法对标准化带来的错误不太敏感。偏离度可以使用glmpca软件包进行计算。
降维。降维旨在减少数据中独立维度的数量。如果不同的基因受同一生物学过程的影响,它们的表达就会存在相关性,这使得降维是可行的。因此,我们不需要单独存储每个基因的信息,而是可以将多个基因的信息压缩成一个特征存储。降维方法在保留有数据集中最有意义的信息结构基础上实现了数据的降维。降维的一个额外好处是降低了噪音,它可以把多个基因(比如,跟某一个通路相关的基因)用类似平均值的操作整合在一起,获得的特征可以反应更精确的表达变化模式。降维后下游分析中的计算工作也减少了,因为只需要针对几个维度而不是数千个基因进行计算。效果更好的降维方案(aggressive dimensionality reduction schemes)可以在二维或三维空间对数据进行可视化以帮助解释结果。
scRNA-seq数据降维的常见第一步是主成分分析(PCA)。PCA在高维空间中鉴定可捕获数据变异最大的轴(也成为主成分,PC)(PCA主成分分析实战和可视化 附R代码和测试数据)。前几个主成分维度捕获了数据集中主要的异质性的信息,因此可以有效的降维。这利用了PCA成熟的理论特性,即,对于给定的矩阵,由前几维PC形成的低阶近似矩阵是原始数据的最佳表示。鉴于此属性,使用前几维PC(或任何类似的低秩近似表示)执行的计算(诸如聚类之类的下游分析)将充分利用数据压缩和去噪的优势。
无论采用哪种方法,用于可视化的降维必然涉及信息丢失并改变细胞之间的距离。因此,直接分析用于绘图的低维坐标是不明智的。相反,这些图应仅只用于解释或传达基于更精确的、更多维度的定量分析结果。这样可以保证分析充分利用了压缩到二维空间时丢失的信息。假如二维图上呈现的细胞分布与使用更多数目的PC进行聚类获得的结果之间存在差异,应倾向于相信后者的结果。
SingleCellExperiment类具有一个专用存储空间reducedDims用于存储降维后的数据(图5.2).scater 软件包提供了多个用于降维分析的便捷函数,可以进行主成分分析(PCA),t-SNE(t-Distributed Stochastic Neighbor Embedding,以及UMAP (Uniform Manifold Approximation and Projection)分析。density包提供了Diffusion map降维方法。zinbwave和glmpca 程序包分别使用零膨胀(zero-inflated)负二项模型和多项式模型进行基于模型的降维分析,优势是在模型中可以考虑混杂因素的影响。
数据整合。由于技术限制(logistical constraints),大型scRNA-seq项目通常需要分多个批次生成数据。但是,不同批次的处理通常会遇到无法控制的差异,例如操作员操作独特性或试剂质量的差异。这导致在不同批次的细胞中观察到的表达发生系统性差异。此外,随着scRNA-seq数据的普及和参考数据集的普及,在整合分析中不可避免地会遇到这种混杂变量的影响。在这个情况下,批次效应可能是数据异质性的主要驱动力,会掩盖相关的生物学差异并使结果的解释变得复杂。
尽管可以使用广义线性模型来整合不同的数据集,但在scRNA-seq分析中,这些方法可能不是最佳的。因为它们基于一个假设,即不同批次的细胞中细胞群体的组成是已知的或相同的。为了克服这一限制,研究者开发了不基于细胞群体构成的先验知识的特制方法用于单细胞数据的批次校正 。这便利了scRNA-seq数据的探索性分析,因为这些先验知识通常是不可用的。
在批次校正之前,最好先检查是否有批次影响。基于特征基因的对数表达值进行PCA分析,再使用基于图的聚类方法展示群体结构。理想情况下,每个聚类簇都应包含来自各个重复scRNA-seq数据集的细胞。然而,如果细胞簇只包含单个批次的细胞,则表明批次效应把本来相同类型的细胞错误地区分开了。诸如t-SNE和UMAP之类的方法也会显示出来自不同批次的细胞之间的差异,这与聚类结果是一致的。值得注意的是,如果某个批次确实包含独特的细胞亚群时,这种依赖于混合程度的可视化诊断可能并不有效,但是仍然是有用的近似方法。
诸如scMerge、scamap之类的包可以使用先验细胞标记(请参阅“注释”部分)进行有监督的整合分析,用以指导对基因表达值进行任何批次校正或调整较低维度的展示形式。另一方面,诸如相互最近邻居(MNN,mutual nearest neighbours)之类的无监督方法会从彼此相邻的邻居集合中识别不同批次中成对的细胞。然后,MNN对中的细胞之间的差异可以用作批次效应的估计值,将其相减得出批处理校正值。实际上,通过调整最近邻居的数量值k,可以调整批次校正的强弱,其中,较高的k值会导致批次之间子群体的匹配更加广泛(generous matching)。这种基于MNN的方法在batchelor软件包中有实现。
批次校正的成功取决于生物异质性信息的保留,因为可以设想一种校正方法将所有细胞简单地聚集在一起,虽然实现了细胞的完美混合,但丢弃了感兴趣的生物信息。为此,CellMixS软件包可用于评估批次之间的细胞混合程度。另一个有用的评估方法是将数据合并后的聚簇结果与每个批次数据分别获得的聚簇结果相比较。理想情况下,我们应该看到多对一的映射关系,跨批次聚簇结果嵌套在批次内聚类结果,这表明任何批次内结构都在校正后得以保留。统计量如兰德指数(Rand index,https://en.wikipedia.org/wiki/Rand_index)可用于评估聚类结果(兰德指数越大聚类效果越好)。
下游统计分析
因研究目标或实验手段的不同,下游分析的方法和工作流程的选择也差异很大。数据前期处理后,可以使用Bioconductor中能够处理SingleCellExperiment类并且可以处理大量细胞的工具进行具体的生物探索分析。我们的在线图书(https://osca.bioconductor. org)为用户提供了用于下游分析和可视化的分析流程和案例研究(图4)。
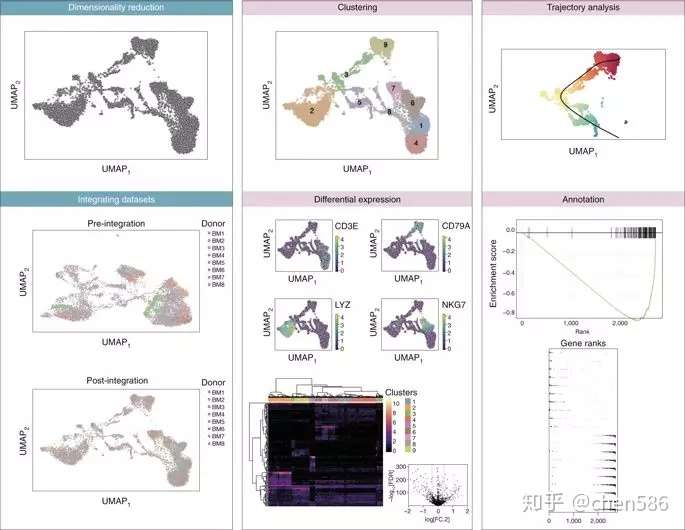
Fig. 4 | Select visualizations derived from various Bioconductor workflows. Various visualizations associated with pre-processing (blue boxes) and downstream statistical analyses (pink boxes). The example data set used throughout was generated as part of the Human Cell Atlas 21 . Details on the generation of these figures are described in our online companion book (https://osca.bioconductor.org).
聚类。在scRNA-seq数据分析中使用经验性的聚类方式定义具有相似表达谱的细胞为一簇。这使我们可以用更容易理解的离散标记来描述种群异质性,而不是试图理解细胞自身所处的高维流形。在基于差异表达获得的标记基因进行注释后,可以将簇视为更抽象的生物学概念(例如细胞类型或状态)的代名词。
值得强调的是细胞簇与细胞类型之间的区别。前者是一种经验称谓,而后者是一个生物学事实(尽管定义有些模糊)。因此,需要认识到聚类,其实像显微镜一样,只是探索数据的一个工具。更改聚类参数可以类比于放大和缩小分辨率来调整观察的粒度,并尝试使用不同的聚类算法来获得数据的其它查看角度。
基于图的聚类方法是对大型scRNA-seq数据集进行聚类分析的一种灵活且扩展性强的技术。在一个高维空间中,每个点(也就是一个细胞)与其最近的邻居相连构成一幅网络图。边基于相连的细胞的相似性加权,连接越相似的细胞的边的权重越高。louvain和leiden等算法 可以用来鉴定细胞簇。
BiocNeighbors提供了用于精确和近似最近邻检测的分析工具,并通过scran构建实际连接图形。值得注意的是,对于大型scRNA-seq数据集,近似NN方法以可接受的准确性损失为代价极大地缩短了运行时间,并具有平滑噪声和稀疏性的额外优势。替代方法包括SIMLR软件包,它使用多个kernal来学习最适合数据的细胞距离度量方式,并可用于聚类和降维。对于大数据,mbkmeans软件包实现了k-means算法的高速版本。最后,SC 和clusterExperiment程序包构建了基于多重参数的一致性聚类比较分析。
许多这些程序包都可以对聚类结果进行定量和视觉评估,此外,还专门设计有用于数据可视化和评估的其它程序包(例如clustree)。另外可以通过一些度量参数(例如簇模块性或轮廓系数silhouette coefficient)来独立评估聚类结果。
差异表达。差异基因表达(DGE)分析可用于识别驱动簇分离的标记基因。这些标记基因使我们能够根据其功能注释为每个簇赋予生物学意义。在最明显的情况下,每个簇的标记基因与已经注释的特定细胞类型相关,从而让聚类结果等同于细胞类型鉴定结果。同时还可以应用相同原理检测更细微的差异,例如激活状态或分化状态之间的比较。DGE分析用于细胞类型注释的替代方案是基因集富集分析,该分析将基因归类到先验的基因模块或生物途径,以便于进行生物解释。我们将在“注释”部分中讨论此主题。
在差异表达方法中,有两种通用方法很突出。第一种方法是把最初广泛应用于普通转录组测序的R包(如edgeR,DESeq2和limma-voom)等通过各种方法(例如通过创建伪普通转录组图谱)改造后应用于scRNA-seq分析。或者,诸如zinbwave之类的方法在离散度估计和模型拟合步骤中减轻在scRNA-seq数据中大量零的权重,然后再进行差异分析,也可以促进普通转录组差异基因分析方法应用于scRNA-seq数据。第二类方法是专门针对单细胞数据的特征开发的,其使用的统计方法直接对scRNA-seq数据常见的大量零值直接建模。这些方法将基因表达明确地分为两个部分:离散部分(描述零与非零表达的基因的比例)以及连续部分(基因表达定量水平)。尽管本文提到的所有方法都可以对”连续部分”进行差异分析,但是只有第二类方法可以明确地对“离散部分”进行建模(explicitly model),从而对表达频率的差异进行统计分析。为此,MAST软件包使用了hurdle model( Hurdle模型是二分类模型与零截尾模型的联合,它可通过对两部分分别进行极大似然估计而得到参数估计值。),而scDD,BASiCS和SCDE 分别使用贝叶斯混合和层级模型。这些方法可以提供更广泛的检测功能,并且可以直接用于SingleCellExperiment类中包含的scRNA-seq数据。
有关DE分析和上述各种软件包的比较分析的更多详细信息,请参见参考资料65–67.
轨迹分析。细胞异质性还可以建模为一个连续的生物过程,如细胞分化。轨迹分析(或伪时间推断)是专门针对单细胞降维分析的一个特殊应用,它使用系统发育方法来沿着(通常是时间连续性的)轨迹对细胞进行排序,如随时间的发育。推断的轨迹可以识别细胞状态之间的过渡、分化过程或动态细胞过程中导致的二分事件。
轨迹推断的最新方法的改进在最大程度地减少了用户输入参数,并且可以基于各种拓扑结构进行差异基因表达分析(例如Monocle,LineagePulse和switchde)。此外,用于轨迹推断的多个Bioconductor软件包(例如,slingshot, TSCAN,Monocle, cellTree和MFA)最近被证明具有出色的性能。由于对于同一个数据集,不同的方法可能产生截然不同的结果,因此一系列的方法和参数设置需要进行比较测试以评估其鲁棒性。(NBT|45种单细胞轨迹推断方法比较,110个实际数据集和229个合成数据集)
Bioconductor通过提供标准化的数据形式(例如SingleCellExperiment类对象)来方便此类测试。参见(74)获得进一步讨论。
聚类簇注释
scRNA-seq数据分析中最具挑战性的任务可以说是聚类簇注释。获得细胞簇方法非常直接,但是要确定每个簇代表的细胞类型或细胞状态则更加困难。完成这个工作需要弥合当前数据集和先验生物学知识之间的鸿沟,而后者并不总能以一致和定量的方式获得。因此,对scRNA-seq数据的注释通常是手动的,并且是分析流程中的常见瓶颈。
为了加快此步骤,可以应用各种计算方法利用先验信息为新的scRNA-seq数据集赋予生物意义。先验信息的最明显来源是与特定生物学过程相关的认证基因集(例如,来自基因本体论(GO,gene ontology)或KEGG通路信息)。另一种方法是将表达谱与已发布的经过领域专家做过注释的参考数据集直接进行比较。
基因集富集。经典基因集富集(GSE)方法的优点是不需要参考表达值。当处理来自文献或其他定性形式的生物学知识的基因集时,这特别有用。在细胞注释时,通常在一组细胞(或簇)上执行GSE分析以识别这些细胞富集的基因集或生物通路。然后可以根据富集的通路推导细胞类型(或状态)。
Bioconductor提供了专用软件包从数据库(如MSigDB、KEGG、Reactome、GO)中获得预定义的基因特征信息。EnrichmentBrowser简化了从此类数据库收集基因集的过程。最初为普通转录组数据开发的基因集富集分析方法也可应用于scRNA-seq数据中特定基因模块的富集。EnrichmentBrowser,EGSEA和fgsea软件包分别提供了一些经典GSE分析的工具。在MAST、AUCell和slalom中也有进行GSE分析的方法。
自动注释细胞。从概念上讲,最直接的注释方法是将单细胞表达谱与先前注释的参考数据集进行比较。然后,根据最相似的参考样本或某些其他相似性指标,将生物标签分配给待确认的细胞。这是一个常见的分类问题,可以通过标准的机器学习技术如随机森林和支持向量机来解决。任何公开且带有标签的RNA-seq数据集(普通或单细胞的)都可以用作参考,其可靠性在很大程度上取决于给参考集细胞进行注释的原始作者的专业性。
SingleR方法提供了一种用于细胞类型注释的自动化系统。SingleR基于具有最高Spearman相关性的参考样本标记细胞,因此可以认为是k-近邻分类的排序变体。为了减少噪声,SingleR可以识别两组细胞之间的标记基因,并仅使用那些标记基因来计算相关性。程序包中包含许多内置参考数据集,这些数据集来自多个项目,包括免疫基因组计划(ImmGen),ENCODE和免疫细胞表达数据库(DICE)。
分析工具易用性 Accessible analysis
随着对单细胞测序数据的分析兴趣日益浓厚,Bioconductor不仅开发了分析数据的方法和软件,而且还优先考虑使数据本身和数据分析工具更易于用户和开发人员使用。具体而言,社区提供了数据包,其中包含公开可用的已发布数据和模拟数据,以及交互式数据可视化工具。这样可以使单细胞数据和数据分析工具更易于访问,使研究人员可以在自己的工作中利用这些资源并使数据分析民主化(democratizes data analysis)。
基准测试。随着新的单细胞检测、统计方法和相应软件的开发,方便数据集的发布、再现现有分析以及实现新工具与现有工具的比较变得越来越重要。Bioconductor收集了一系列数据包,着重于提供可以直接用于分析的带有版本信息的数据,以及可用于复制手稿图形和展示数据特征的手册。
为了便于查询Bioconductor上已发布的数据包,ExperimentHub包允许使用标准化接口以编程方式访问已发布的数据集。值得注意的是,scRNAseq软件包可以从各种来源获得校正过的高质量scRNA-seq数据集。另外,模拟数据集对评判软件也很有帮助。
另外,splatter包可以模拟包含多种细胞类型、批次效应、不同水平的drop-out事件、差异基因表达和轨迹的模拟scRNA-seq数据集。splatter 包使用自己的模拟策略框架,并整合其它不同模型的模拟策略以提供全面的单细胞模拟数据资源。
为了提高评估单细胞方法性能的基准比较的可重复性,Bioconductor开发了存储不同方法比较结果的基本架构。SummarizedBenchmark 和CellBench软件包提供了用于存储元数据(方法参数和软件包版本)和评估指标的接口。
交互式数据可视化。网络技术的成熟为交互式数据探索开辟了新的途径,而R包shiny则有助于开发丰富的图形用户界面。iSEE和singleCellTK软件包为通过Internet浏览器对scRNAseq数据集进行交互可视化提供了全功能的应用程序,消除了对编程经历的需求。如果实例托管在Web上,则无需编程经验。这两个软件包都直接与SingleCellExperiment数据容器连接以便促进scRNA-seq分析结果的交流。
展望
自从基因组学问世以来,Bioconductor项目就已经通过R统计编程语言拥抱了开源和开放软件的开发。Bioconductor已建立协调包版本和代码审查的最佳实践。除了社区贡献的软件包,核心开发团队(https://www.bioconductor.org/about/core-team)开发并维护必要的基础架构,并审核提交的软件包,以确保它们满足一套指导原则和保证各个软件包之间的兼容性。这些软件包被组织到`BiocViews`中,一个按任务或技术对软件包进行分类的主题注释库。例如,单细胞分析主题在视图`SingleCell`下标记。最重要的是,更广泛的Bioconductor社区(包括论坛、Slack或邮件列表)是代码共享和技术帮助中无私的典范。这些实践共同产生了高质量、维护良好的软件包,为生物学研究提供了一个统一而稳定的分析环境。
最近,Bioconductor社区开发了最新的计算方法、数据结构和交互式数据可视化工具用于分析从单细胞实验中获得的数据。新兴的单细胞技术,包括表观基因组学、T细胞和B细胞文库、空间转录组谱分析和基于测序的蛋白质谱分析,希望能推动计算生物学的发展。特别是,支持多组学分析的技术正在迅速发展,Bioconductor为研发用于此类技术分析的统计方法奠定了必要的基础。
此外,Bioconductor的标准化数据容器可实现Bioconductor软件包以及与其他软件之间的互操作性。可以将存储在SingleCellExperiment中的数据转换为Seurat、Monocle 和Python的scanpy可用的格式,从而可以使用最能满足当前分析目标的工具。实际上,R与其他编程语言有着很长的互操作性历史。有四个例子,Rcpp用于将C++编译后的代码集成到R软件包中,rJava软件包用于从R中调用Java代码的,R中的.Fortran()函数可以调用Fortran代码,reticulate包与Python互通。
这种互操作性使常见的机器学习框架(例如TensorFlow/Keras)可以直接在R中使用。
对于新手来说,Bioconductor中能进行大量单细胞分析的数量众多的程序包可能令人望而生畏。为了解决单细胞分析中越来越多软件包的选择问题,我们总结并强调了当前最先进的数据基础架构、方法和软件,并按照典型的单细胞分析流程组织了这些软件包(图3)(图4)。最后,我们开发了在线的配套书籍,其中提供了有关各个分析主题的更详细信息以及完整的代码流程(https://osca.bioconductor.org)。随着新软件包的出现,我们会不断更新和维护这套在线书籍,促进Bioconductor资源更方便使用。
作者:张虎
编辑:生信宝典
单细胞系列教程
参考文献
50. Andrews, T. & Hemberg, M. M3Drop: Dropout-based feature selection for scRNASeq. Bioinformatics 35, 2865–2867 (2019).
51. Yip, S. H., Sham, P. C. & Wang, J. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Brief. Bioinform. 20, 1583–1589 (2018).
65. Soneson, C. & Robinson, M. D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018).
66. Wang, T., Li, B., Nelson, C. E. & Nabavi, S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinform. 20, 40 (2019).
67. Crowell, H. L. et al. On the discovery of population-specific state transitions from multi-sample multi-condition single-cell RNA sequencing data. Preprint at bioRxiv https://doi.org/10.1101/713412 (2019).
74. Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547 (2019).
https://blog.sciencenet.cn/blog-118204-1220242.html
上一篇:重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)
下一篇:众志成城 - 财经、医学、数学、古脊椎多领域团队19文解析新型冠状病毒 - 起源、传播、防控 - 为决策助力