博文
还原应激和还原损伤
|
氧化应激之还原应激
重要性:还原当量(NAD(P)H和谷胱甘肽[GSH])对于维持细胞氧化还原平衡和调节细胞代谢至关重要。由过量的还原型NAD+(NADH)、还原型NADP+(NADPH)和GSH引起的还原性压力与氧化性压力一样有害,并且与许多病理过程有关。
近期进展:还原性压力拓宽了我们对细胞氧化还原平衡重要性以及氧化还原环境失衡对生物功能影响的视野,包括细胞代谢。
关键问题:细胞内NAD(H)、NADP(H)和GSH/GSSG的分布高度区室化。理解细胞如何在无压力和有压力的条件下协调不同氧化还原对池是全面了解氧化还原平衡和压力的关键。探索还原性压力的潜在机制及其生物学后果,包括对能量代谢的影响,也是至关重要的。
未来方向:未来的研究需要调查还原性压力如何影响细胞代谢以及细胞如何适应还原性压力对其代谢的影响。是否NADH穿梭和线粒体烟酰胺核苷酸转氢酶可以调节缺氧诱导的还原性压力也值得探究。开发策略(例如,抗还原剂方法)以抵消还原性压力及其相关的不良生物后果也需要广泛的未来努力。
Xiao W, Loscalzo J. Metabolic Responses to Reductive Stress. Antioxid Redox Signal. 2020 Jun;32(18):1330-1347.
引言
哺乳动物细胞依赖于一系列氧化和还原(redox)反应来产生能量(例如,ATP)并从营养物质合成必需的细胞组分(例如,核酸),以支持其生物功能。在氧化还原反应中,电子从还原剂(还原物)流向氧化剂(氧化物)。细胞内的氧化还原对,主要是烟酰胺腺嘌呤二核苷酸(NAD+)/还原型NAD+(NADH)、磷酸化NAD+(NADP+)/还原型NADP+(NADPH)和还原型谷胱甘肽(GSH)/GSSG,负责大部分的细胞电子转移。这些氧化还原对作为辅因子或底物,用于酶促或非酶促中和活性氧种(ROS),以在细胞内维持相对还原的环境。
重要的是,这些氧化还原对还将细胞的氧化还原环境与细胞能量学联系起来。例如,NAD+作为电子接收器支持糖酵解;NADH为线粒体氧化磷酸化(OXPHOS)提供电子;NADPH是脂肪酸和核酸还原性生物合成的主要电子来源。鉴于这些还原当量对细胞氧化还原平衡和能量代谢不可或缺,这些分子的氧化还原状态失衡与多种病理条件有关,如心血管疾病、神经退行性疾病、癌症和衰老。
在本综述中,我们首先介绍细胞氧化还原网络和氧化还原压力,然后描述NAD+/NADH、NADP+/NADPH和GSH/GSSG氧化还原对的代谢来源和细胞分布。这些信息之后是对扰动这些比例导致还原性压力的解释。最后,我们讨论了对还原性压力的代谢反应及其对细胞功能的影响。
一、细胞氧化还原网络和氧化还原压力
哺乳动物细胞中的氧化还原网络
细胞的氧化还原环境由促氧化剂和抗氧化剂平衡。细胞内的促氧化剂包括活性氧种(例如,超氧阴离子[O2•−]和过氧化氢[H2O2])、活性氮种(例如,一氧化氮[NO•])及其衍生物(例如,过氧亚硝酸盐[ONOO−]),它们主要由酶产生,例如NAD(P)H氧化酶(NOXs)和解偶联型一氧化氮合酶,或作为功能性副产品由线粒体电子传递链产生。为了抵消这些氧化剂,细胞发展了抗氧化系统,包括抗氧化酶,例如超氧化物歧化酶(SOD1–3)、过氧化氢酶、谷胱甘肽过氧化物酶(GPx1–8)、硫氧还蛋白(Trx1–2)和过氧化物还原酶(Prx1–6);和非酶小分子,例如GSH、α-生育酚和抗坏血酸(7, 30, 87)(图1)。
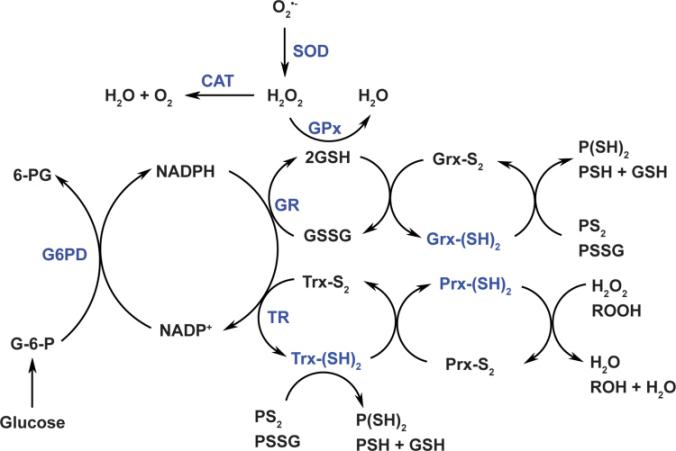
图1细胞氧化还原网络。细胞氧化还原平衡是通过促氧化剂和抗氧化剂系统之间的微妙平衡来维持的。细胞内的促氧化剂主要包括O2•−和H2O2,它们是由酶产生的,或者/和通过线粒体呼吸作用产生。SODs将O2•−转化为H2O2,然后H2O2被过氧化氢酶、GPxs和Prxs进一步中和成水。过氧化氢酶对H2O2的亲和力很低(但转化率高,催化效率高),并且当H2O2水平达到毫摩尔范围时,可以将其还原成水。GPxs需要两个分子的还原型GSH来还原H2O2,而GSH在此反应中被氧化成GSSG。GSH也可以被谷胱甘肽还原酶[Grx-(SH)2]用来还原蛋白质内/间二硫键(PS2或PSSG)成为还原的蛋白质半胱氨酸残基(P[SH]2或PSH + GSH)。GSSG还原成GSH的过程是由GR催化的,GR使用NADPH作为电子供体。Prxs从还原型硫氧还蛋白[Trx-(SH)2]中提取电子来还原H2O2或有机过氧化物(ROOH)成水或/和醇(ROH),同时Trx-(SH)2被氧化成Trx-S2。与GSH类似,Trx-(SH)2也可以还原蛋白质二硫键成蛋白质半胱氨酸。Trx-S2的还原过程是由TRs催化的,使用NADPH作为电子供体。NADPH是通过葡萄糖代谢的磷酸戊糖途径(PPP)中的G6PD产生的。因此,细胞的氧化还原状态和细胞代谢通过NADPH紧密联系。改编自参考文献(7, 8)并获许可。6-PG,6-磷酸葡萄糖酸;G-6-P,葡萄糖-6-磷酸;G6PD,葡萄糖-6-磷酸脱氢酶;GPxs,谷胱甘肽过氧化物酶;GR,谷胱甘肽还原酶;Grx,谷胱甘肽还原酶;GSH,谷胱甘肽;GSSG,GSH二硫化物;H2O2,过氧化氢;NADPH,还原型NADP+;O2•−,超氧阴离子;PPP,磷酸戊糖途径;Prxs,过氧化物还原酶;PSSG,蛋白质-谷胱甘肽二硫化物;SODs,超氧化物歧化酶;TRs,硫氧还蛋白还原酶;Trx,硫氧还蛋白。
值得注意的是,细胞氧化剂的产生是特定于区域的,但它们的目标可以是局部的或/和远离源头区域的。例如,线粒体产生的H2O2可以在线粒体内(局部)或/和细胞质中(远程)作为信号分子发挥作用(26)。在血管系统中,内皮细胞衍生的NO•可以从内皮扩散到相邻的平滑肌细胞,导致血管舒张(22)。与氧化剂不同,抗氧化酶根据它们特定的亚细胞定位发挥功能。例如,位于线粒体基质的锰SOD(SOD2)只能在线粒体中将O2•−转化为H2O2,然后H2O2被线粒体过氧化物酶(如过氧化氢酶、GPx1、GPx4和Prx2)进一步还原成水。
GSH/GSSG对偶和NADP+/NADPH对偶在过氧化物的两电子还原中发挥关键作用。GSH是GPxs清除H2O2的共底物。在这种情况下,两个分子的GSH捐赠两个电子来将H2O2还原成水,并将自己氧化成GSSG,然后GSSG通过谷胱甘肽还原酶(GR)使用NADPH作为电子供体循环回GSH(7, 8)(图1)。有趣的是,谷胱甘肽还原酶(Grx1–2)也利用GSH来还原蛋白质内/间二硫键(PS2或蛋白质-谷胱甘肽二硫化物[PSSG])成蛋白质巯基团(-SH)(7, 8)(图1)。除了GR外,NADPH还是硫氧还蛋白还原酶(TRs)不可或缺的辅因子。同样地,NADPH为TRs提供两个电子来将氧化的Trx(Trx-S2)还原为其活性二硫醇形式Trx-(SH)2。还原型Trx-(SH)2作为Prxs再生的电子来源,分别将H2O2和其他有机过氧化物还原成水和醇(7, 8)(图1)。值得注意的是,牛组织中总Trx含量估计为1–10 μM,比总GSH水平(mM范围)低2–3个数量级。
氧化还原压力
在生理条件下,细胞促氧化剂保持在许多生物过程所需的稳态水平,如细胞信号传导、增殖和分化(30, 87)。然而,当促氧化剂和抗氧化剂之间的平衡受到不利扰动时,包括氧化性压力和还原性压力在内的氧化还原压力就会发生(图2)。
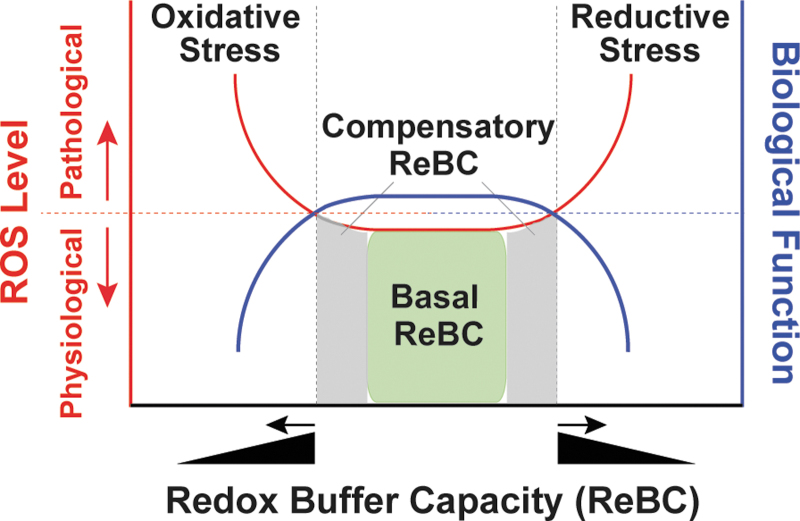
图2. 氧化还原压力。NADH/NAD+、NADPH/NADP+和GSH/GSSG氧化还原对是主要的细胞氧化还原缓冲剂。在无压力条件下,这三个氧化还原对有足够的能力维持氧化还原平衡,称为基础ReBC(X轴;红线下的绿色框)。当细胞ROS水平增加时(左Y轴;红线),这些氧化还原缓冲剂也能通过提升基础ReBC到一定水平来做出响应,称为补偿性ReBC(X轴;红线下的灰色区域)。在这两种情况下,细胞ROS水平都保持在生理水平以确保正常的生物功能,如信号分子的充足性(右Y轴;蓝线)。然而,当这种补偿响应持续超过一定阈值,即最大ReBC被细胞还原ROS水平超过时(X轴,黑三角),就可能发生还原性压力(红线)。相比之下,当细胞ReBC降低(X轴,黑三角),或/和细胞氧化ROS产生的压倒性时,就会发生氧化性压力。还原性压力和氧化性压力(统称为氧化还原压力)都能促进ROS的产生,导致大分子的氧化损伤和细胞功能的紊乱。NAD+,烟酰胺腺嘌呤二核苷酸;NADH,还原型NAD+;NADP+,磷酸化NAD+;ReBC,氧化还原缓冲能力;ROS,活性氧物种。
氧化性压力的概念最初是由Paniker等人在1970年研究H2O2刺激的正常和GR缺陷型人类红细胞中的GSH/GSSG对时提出的(74)。这个术语现在被定义为细胞促氧化剂水平和抗氧化能力之间的不平衡,偏向前者(30, 52, 87),意味着过多的氧化剂水平会导致氧化性压力(图2)。普遍接受的是,由过量ROS/RNS水平引起的氧化性压力对生物功能有害,并且与许多病理条件的发病机制有关,例如心血管疾病、神经退行性疾病、衰老和癌症(30, 90, 106);然而,重要的是要注意,这些氧化剂(例如,O2•−和H2O2)能够根据两个反应物的标准氧化还原电位(E°′)氧化或还原其他分子(表1)(89)。例如,在芬顿反应中,O2•−被氧化成O2,H2O2被还原成羟基自由基(HO•),这是最具氧化性的物种(E°′ = 2310 mV)。
O2•− + Fe3+ → O2 + Fe2+ (1)
H2O2 + Fe2+ → Fe3+ + HO• + HO− (2)
O2•− + H2O2 → O2 + HO• + HO− (Net reaction)
同样,H2O2能够还原高铁血红蛋白并氧化蛋氨酸;而它本身则分别被氧化成O2•−和被还原成水(52)。因此,O2•−和H2O2水平的增加可以看作是根据氧化还原偶联物种的相对丰度,产生氧化性或还原性压力。
与氧化性压力不同,还原性压力并不是一个定义得很清晰的概念。这个术语最初是由Gores等人在1989年描述的,实验中作者通过用化学药品处理大鼠肝细胞来模拟缺氧,以阻断线粒体呼吸和ATP产生(所谓的化学缺氧)(27)。作者提出,由于氧气供应有限(例如,缺氧),电子载体变得还原,并在重新供氧时(例如,再灌注)被重新氧化,导致ROS产生的爆发,称为“还原性压力”(27)。这个术语目前有一个更一般化的定义,即还原性压力是细胞促氧化剂水平和还原能力之间不平衡,偏向后者(30, 52, 87)。在本综述的背景下,我们将还原性压力定义为还原当量(特别是NADH、NADPH和GSH)的过量积累,超过了内源性氧化还原酶的能力。
在生理条件下,细胞氧化还原缓冲剂[GSH/GSSG和NAD(P)H/NAD(P)+]有足够的能力(称为基础氧化还原缓冲能力[ReBC])将细胞氧化剂和还原剂维持在生理水平(ROS作为信号分子)(图2)。当细胞承受氧化性或还原性损伤时,氧化还原缓冲剂增加到一定水平(称为补偿性ReBC)以抵消这些氧化还原压力并恢复氧化还原平衡。在这些情况下,细胞氧化剂和还原剂仍然维持在生理范围内。然而,当这种补偿响应达到最大值时,ReBC被超过,就会发生氧化性或还原性压力(图2)。重要的是,还原性压力将细胞ROS水平降低到低于生理水平,从而干扰它们的信号功能。从不同的角度来看,还原性压力也可以促进ROS的产生(例如,通过部分还原氧气),因此本质上被认为促进了“氧化性压力”,这取决于这些ROS参与的氧化还原对(45, 92, 107)。
像氧化性压力一样,还原性压力也会损害细胞功能(31)。例如,内质网(ER)具有相对氧化的环境,这对于膜和分泌蛋白的结构二硫键形成是必需的。然而,在还原性压力下,蛋白质二硫键不恰当地形成,导致未折叠蛋白反应和ER应激的激活(60)。与此概念一致,我们和其他研究表明,还原性压力干扰了蛋白质二硫键的形成,导致酵母中的ER应激和人类细胞中膜蛋白的胞内滞留(32, 60, 100, 113)。此外,心脏特异性过表达突变的人类αB-晶状体蛋白(hR120GCryAB)的小鼠显示出GSH水平升高以及GR、葡萄糖-6-磷酸脱氢酶(G6PD)、过氧化氢酶和GPx活性的增加(表明存在还原性压力),这导致了这些小鼠心脏肥大和心力衰竭的发展(77, 78)。在早期糖尿病动物中观察到细胞质NADH/NAD+的升高及其导致的还原性压力,这是由于底物(例如,山梨醇、非酯化脂肪酸和葡萄糖醛酸途径代谢物)的氧化增加,这与NAD+还原为NADH相耦合(38)。因此,还原性压力对细胞是有害的,并且与许多病理条件有关,包括心血管疾病和糖尿病(31, 51, 105)。
二、代谢来源和细胞分布的NAD(H)和NADP(H)对
NAD+是许多酶(包括代谢酶)的关键辅助因子,对各种生物过程至关重要(106)。哺乳动物细胞可以从其前体物质合成NAD+,并从NADH循环回收NAD+。根据前体的可用性,可以通过三条途径合成NAD+:使用色氨酸为前体的全新途径,使用烟酸为前体的Preiss-Handler途径,以及使用烟酰胺(NAM)和烟酰胺核糖苷(NR)为前体的回收途径(106)。NAD+可以通过NAD+激酶(NADKs)磷酸化成NADP+;NAD+和NADP+的氧化形式可以通过相应的脱氢酶还原成NADH和NADPH(106)。我们和其他研究者已经广泛回顾了所有三个NAD+生物合成途径的酶促步骤和调节因素(106, 112, 114)。在这篇综述中,我们主要关注通过代谢酶进行的NAD(H)和NADP(H)之间的相互转化。
NAD(H)和NADP(H)对的代谢来源
NAD(P)+与NAD(P)H之间的相互转化是由参与糖酵解、磷酸戊糖途径(PPP)和三羧酸循环(TCA循环)的代谢酶介导的(图3和4.4)。糖酵解和TCA循环是细胞质和线粒体NAD(H)的两个主要来源(图3)。在细胞质中,NAD+与NADH之间的相互转化由两种酶催化,即甘油醛-3-磷酸脱氢酶(GAPDH)和乳酸脱氢酶(LDH)(56, 106)。GAPDH催化甘油醛-3-磷酸(G-3-P)与1,3-二磷酸甘油酸的可逆转化,同时伴随着NAD+/NADH对的相应转化。LDH在糖酵解中将丙酮酸还原为乳酸,同时将NADH氧化为NAD+。值得注意的是,当乳酸作为TCA循环的燃料源被利用时,LDH也可以催化反向反应,将NAD+还原为NADH并将乳酸氧化为丙酮酸(37, 94)。细胞质中NAD(H)的其他来源包括醇脱氢酶(ADHs)和醛脱氢酶(11)。
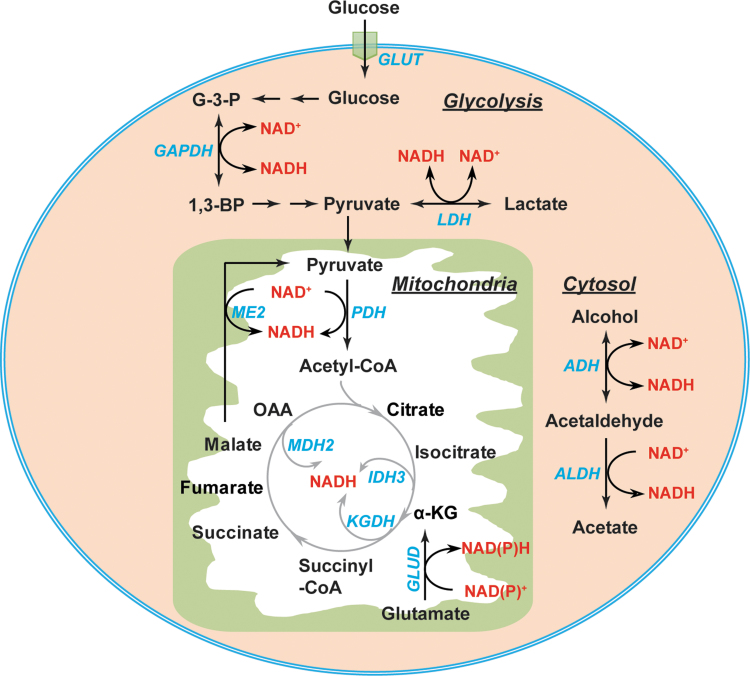
图3. NAD(H)的代谢来源。在细胞质中,NAD+与NADH之间的相互转化由两种酶GAPDH和LDH以及醇代谢酶ADH和ALDH介导。在线粒体基质中,PDH、ME2、GLUD以及TCA循环酶(IDH3、KGDH和MDH2)有助于NAD(H)的产生。1,3-BPG,1,3-二磷酸甘油酸;α-KG,α-酮戊二酸;ADH,醇脱氢酶;ALDH,醛脱氢酶;G-3-P,甘油醛-3-磷酸;GAPDH,甘油醛-3-磷酸脱氢酶;GLUD,谷氨酸脱氢酶;GLUT,葡萄糖转运蛋白;IDH,异柠檬酸脱氢酶;KGDH,α-酮戊二酸脱氢酶;LDH,乳酸脱氢酶;MDH2,苹果酸脱氢酶2;ME,苹果酸酶;OAA,草酰乙酸;PDH,丙酮酸脱氢酶;TCA,三羧酸。
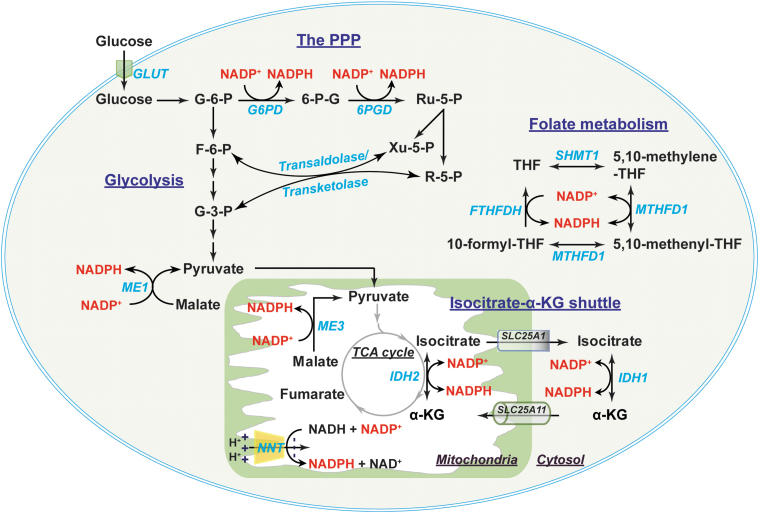
图4.细胞内NAD(H)和NADP(H)的代谢来源及分布
在细胞质中,NAD+与NADH之间的相互转化由两种酶GAPDH和LDH以及醇代谢酶ADH和ALDH介导。在线粒体基质中,PDH、ME2、GLUD以及TCA循环酶(IDH3、KGDH和MDH2)有助于NAD(H)的产生。1,3-BPG, 1,3-二磷酸甘油酸;α-KG, α-酮戊二酸;ADH, 醇脱氢酶;ALDH, 醛脱氢酶;G-3-P, 甘油醛-3-磷酸;GAPDH, 甘油醛-3-磷酸脱氢酶;GLUD, 谷氨酸脱氢酶;GLUT, 葡萄糖转运蛋白;IDH, 异柠檬酸脱氢酶;KGDH, α-酮戊二酸脱氢酶;LDH, 乳酸脱氢酶;MDH2, 苹果酸脱氢酶2;ME, 苹果酸酶;OAA, 草酰乙酸;PDH, 丙酮酸脱氢酶;TCA, 三羧酸。
在细胞质中,NADPH主要由PPP途径中的G6PD和6PGD产生,其中产物R-5-P和Xu-5-P可以通过转醛酶和转酮酶转入糖酵解途径。ME1和IDH1也有助于细胞质NADP(H)的产生。此外,叶酸代谢中的两个酶MTHFD1和FTHFDH也是细胞质NADP(H)的来源。线粒体NADP(H)由NADP+依赖的IDH2、NNT和ME3产生。细胞质和线粒体的NADP(H)池通过异柠檬酸-α-KG穿梭交换,其中细胞质IDH1和线粒体IDH2催化异柠檬酸和α-KG的互变以及NADP+和NADPH的互变。柠檬酸载体蛋白(由SLC25A1基因编码)和α-KG/苹果酸反向运输蛋白(由SLC25A11基因编码)分别介导异柠檬酸和α-KG在细胞质和线粒体之间的运输。6PGD, 6-磷酸葡糖酸脱氢酶;FTHFDH, 10-甲酰-四氢叶酸脱氢酶;MTHFD, 甲基-四氢叶酸脱氢酶;NNT, 烟酰胺核苷酸转氢酶;R-5-P, 核糖-5-磷酸;Ru-5-P, 核糖-5-磷酸;SCL25A1, 溶质载体家族25成员1;SCL25A11, 溶质载体家族25成员11;SHMT, 丝氨酸羟甲基转移酶;THF, 四氢叶酸;Xu-5-P, 木酮糖-5-磷酸。
在线粒体中,每分子葡萄糖可以通过TCA循环在氧气充足的条件下产生八分子NADH。丙酮酸脱氢酶(PDH)复合体在将丙酮酸脱羧为乙酰辅酶A的同时将NAD+还原为NADH。后者进入TCA循环,在IDH3、KGDH和MDH2(56, 106)(图3)的作用下产生NADH。此外,与NAD+连接的苹果酸酶(ME2)和谷氨酸脱氢酶(GLUD1–2)也有助于线粒体NADH池的产生(81, 106)(图3)。
细胞质NADPH主要由PPP途径中的G6PD和6-磷酸葡糖酸脱氢酶(6PGD)(56, 106, 112)产生。G6PD将葡萄糖-6-磷酸(G-6-P)转化为6-磷酸葡糖酸,后者进一步被6PGD代谢为核糖-5-磷酸。这两个反应都与NADP+还原为NADPH相偶联(图4)。值得注意的是,核糖-5-磷酸及其衍生物核糖-5-磷酸(R-5-P)可以通过转醛酶/转酮酶通过一系列非氧化反应转化为果糖-6-磷酸(F-6-P)和G-3-P,并由此转入糖酵解途径(56)。
此外,其他酶也有助于细胞质NADPH池的产生,如IDHs、MEs、MTHFDs和FTHFDHs(56, 111),所有这些酶都有细胞质和线粒体同工酶。例如,细胞质IDH(IDH1)和线粒体IDH2催化异柠檬酸氧化为α-酮戊二酸(α-KG)以及NADP+还原为NADPH。细胞质苹果酸酶(ME)1将苹果酸脱羧为丙酮酸,同时将NADP+还原为NADPH。此外,叶酸代谢中的两个酶也产生NADPH;细胞质MTHFD(MTHFD1)催化5,10-甲基-四氢叶酸(5,10-methylene-THF)转化为5,10-次甲基-THF;细胞质FTHFDH催化10-甲酰-THF回收为THF(111)。
线粒体NADPH可以由线粒体酶IDH2、ME3、MTHFD2和FTHFDH通过与其相应的细胞质同工酶相同的反应产生(56, 111)(图4)。NADP+依赖的GLUDs也可以通过将谷氨酸转化为α-KG来产生NADPH(81)。值得注意的是,线粒体NADPH的另一个重要来源是烟酰胺核苷酸转氢酶(NNT)。NNT位于线粒体内膜,利用线粒体内膜两侧的质子梯度作为驱动力,从NADH获得电子以还原NADP+为NADPH:NADH + NADP+ ← → NAD+ + NADPH(35, 84)。据估计,在C57BL/6N小鼠心脏组织中,IDH2、NNT和ME3分别贡献了线粒体NADPH产生的70%、22%和8%(69)。
NAD(H)和NADP(H)的细胞内区室分布
由于生物合成酶的特定定位和这些二核苷酸对膜的通透性,细胞内NAD(H)和NADP(H)的分布高度区室化(106)。例如,线粒体烟酰胺单核苷酸腺苷酰转移酶和线粒体NADK分别支持该器官中的NAD+和NADP+的合成(10, 71, 118)。虽然NAD(P)+可以自由穿过核膜,但这些二核苷酸都不能扩散穿过线粒体内膜(35, 114)。值得注意的是,不同区室的NAD(H)和NADP(H)池对外源刺激的差异响应。通常,细胞质和核NAD(H)池比线粒体NAD(H)池对氧化应激的变化更敏感,而线粒体NAD(H)池稳定维持,对抗氧化损伤是细胞存活所必需的(108, 120)。
尽管细胞质、核和线粒体NAD(H)和NADP(H)池通常在其各自区室内发挥作用,这些区室的二核苷酸池也通过穿梭机制交换,以维持整体细胞的氧化还原环境和依赖氧化还原的功能。细胞质NADH可以自由扩散穿过多孔的线粒体外膜;然而,它无法穿过线粒体内膜(10, 76, 112)。为了克服这一限制,细胞发展了两种NADH穿梭机制:苹果酸/天冬氨酸穿梭和甘油-3-磷酸穿梭(35, 65, 106)。
在第一次穿梭中,细胞质中的NADH被MDH1氧化为NAD+,同时将草酰乙酸酯(OAA)还原为苹果酸;而在线粒体中,NAD+被MDH2还原为NADH,伴随着苹果酸在TCA循环中氧化回OAA。这种穿梭还需要两种转运蛋白,α-KG/苹果酸逆向转运蛋白(由SLC25A11基因编码),它将细胞质中的苹果酸转移到线粒体基质中;以及天冬氨酸/谷氨酸逆向转运蛋白(由SLC25A13基因编码),它将线粒体中的天冬氨酸转移到细胞质中。与苹果酸/天冬氨酸穿梭不同,甘油-3-磷酸穿梭是不可逆的,只需要一种酶,即甘油-3-磷酸脱氢酶(GPDH)。细胞质中的GPDH将NADH氧化为NAD+,同时将二羟丙酮磷酸(DHAP)还原为甘油-3-磷酸,而线粒体中的GPDH催化相反的反应,将甘油-3-磷酸氧化为DHAP,并将FAD还原为FADH2。因此,这样的穿梭机制使得细胞能够在细胞质中维持足够的NAD+以支持糖酵解,以及在线粒体中维持足够的NADH以支持氧化磷酸化。
像NADH一样,NADP(H)也不能跨越线粒体内膜扩散。因此,细胞质和线粒体NADP(H)池的交换是通过异柠檬酸-α-KG穿梭通过IDH1和IDH2同工酶来完成的。细胞质中的IDH1和线粒体中的IDH2催化异柠檬酸和α-KG之间的可逆转化,同时伴随着NADP+和NADPH的互变。这种穿梭还需要两种载体蛋白,即柠檬酸载体蛋白(由SLC25A1基因编码),它将线粒体中的异柠檬酸输出到细胞质中以交换苹果酸;以及α-KG/苹果酸逆向转运蛋白,它是苹果酸/天冬氨酸穿梭中的同一载体。因此,异柠檬酸-α-KG穿梭在维持细胞NADPH水平中起着关键作用。综合来看,这些穿梭机制使得细胞能够在广泛的生理病理条件下维持氧化还原和能量稳态。
三、还原型谷胱甘肽(GSH)的生物合成和细胞分布
GSH是哺乳动物细胞中含量最丰富的硫醇抗氧化剂。它作为H2O2去除的底物,通过GPxs发挥作用,并自身被氧化为GSSG。GSSG还原为GSH的过程是由GR催化的,GR使用NADPH作为电子供体。除了其抗氧化功能外,GSH还参与细胞氧化还原信号的调节(例如,蛋白质S-谷胱甘肽化)、细胞增殖和细胞死亡的调控,以及对异物及其代谢产物的解毒。
GSH的生物合成
GSH是一种三肽,由前体氨基酸l-谷氨酸、l-半胱氨酸和甘氨酸在细胞质中通过两个ATP依赖的步骤合成。在第一步中,谷氨酸与半胱氨酸在限速酶γ-谷氨酰半胱氨酸连接酶(GCL)的催化下形成γ-谷氨酰半胱氨酸。GCL酶由两个功能亚基组成:催化亚基(GCLC)和修饰亚基(GCLM)。在第二步中,GSH合成酶(GS)通过将甘氨酸添加到γ-谷氨酰半胱氨酸上来催化GSH的形成。值得注意的是,谷氨酸和半胱氨酸通过一个非传统的γ-连接肽键连接,这种肽键只能被γ-谷氨酰转肽酶(GGT)水解。由于GGT在特定细胞的外表面表达(例如,肝脏、肺部、血管内皮、肾小管细胞和胆管上皮细胞),GSH的降解仅发生在细胞外空间;细胞内的GSH不易被降解。
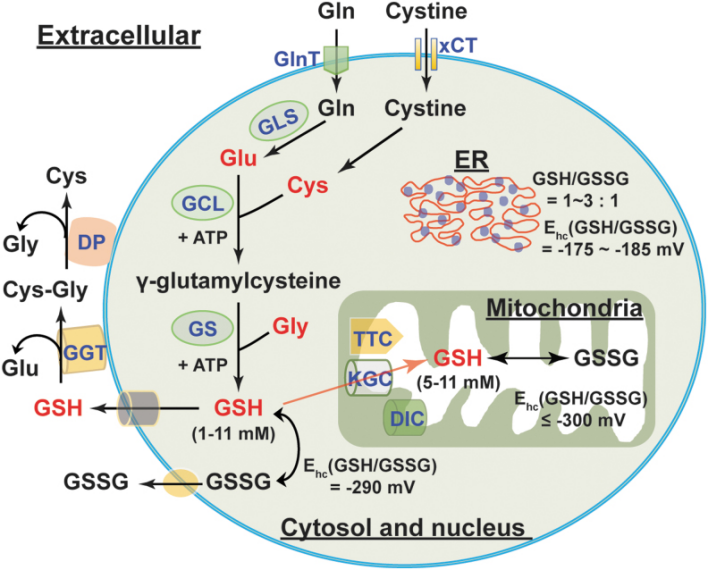
图5生物合成和细胞分布的谷胱甘肽(GSH)。Glu、Cys 和 Gly 是 GSH 的三个前体氨基酸。GSH 的合成需要两个连续步骤,由两个 ATP 依赖性酶催化:GCL(限速酶)和 GS。Glu 可以通过内源性和外源性 Gln 经 GLS 介导的谷氨酰胺分解得到补充。Cys 可以通过还原内源性和外源性胱氨酸生成。细胞质和细胞核中的 GSH 水平在 1-11 mM 范围内,半细胞还原电位(Ehc)为 -290 mV。相比之下,内质网具有更氧化的环境,GSH/GSSG 比例为 1-3:1,Ehc 为 -175 至 -185 mV。细胞质中的 GSH 可以通过三种潜在的转运蛋白 KGC、DIC 和 TTC 运输到线粒体中。线粒体 GSH 水平和 Ehc 与细胞质和细胞核池相当。此外,GSH 和 GSSG 也可以分泌到细胞外 compartment;然而,它们的水平相对较低(在微摩尔范围内)。细胞外的 GSH 可以被细胞表面酶 GGT 降解,形成 Glu 和 Cys-Gly,进一步被 DP 代谢成游离的 Cys 和 Gly。产生的氨基酸可以被细胞重新利用于细胞内 GSH 的合成。Cys,半胱氨酸;Cys-Gly,半胱氨酰甘氨酸;DIC,二羧酸载体;DP,二肽酶;ER,内质网;GCL,γ-谷氨酰半胱氨酸连接酶;GGT,γ-谷氨酰转肽酶;GLS,谷氨酸酶;Glu,谷氨酸;Gly,甘氨酸;GS,GSH 合酶;KGC,α-酮戊二酸载体;TTC,三羧酸载体;xCT,胱氨酸转运蛋白。
GSH 合成的调节取决于其组成氨基酸的可用性,尤其是半胱氨酸和谷氨酸,以及 GCL 的酶活性。Lu 在 2013 年的一篇优秀综述中系统总结了调节半胱氨酸可用性和 GCL 活性的机制(55)。在这篇综述的背景下,我们讨论了最近发现的调节细胞 GSH 水平的机制。
缺氧诱导因子 1α(HIF-1α)是缺氧条件下细胞代谢的中心转录调节因子。充分的证据支持(假)缺氧通过 HIF-1α 依赖性和 HIF-1α 非依赖性机制增加细胞 GSH 水平。Lu 等人报道,在缺氧条件下,三阴性乳腺癌细胞中 GCLM 亚基和胱氨酸转运蛋白(xCT,由 SLC7A11 基因编码)是 HIF-1α 的转录靶标(53)。用化疗药物治疗这些细胞可刺激这两个基因的 mRNA 和蛋白质表达,并通过 HIF-1α 依赖性机制增加细胞内 GSH 水平(53)。敲除含有脯氨酸羟化酶结构域蛋白 2(PHD2)稳定了 HIF-1α 蛋白并激活了 HIF-1α 信号,这在转录上上调了谷氨酰胺酶 1(GLS1)的表达,导致 ROS 产生减少,细胞 GSH 水平、GSH/GSSG 增加,以及小鼠骨膜祖细胞的存活率提高,这表明 HIF-1α 信号促进了谷氨酰胺分解以提供谷氨酸用于 GSH 合成(96)。
有趣的是,在缺氧条件下也报道了 HIF-1α 非依赖性的 GSH 合成调节。最近的研究表明,缺氧上调了 xCT SLC7A11 的表达,并增加了多种癌细胞中的细胞内胱氨酸水平和 GSH 合成(62, 82)。从机制上讲,SLC7A11 的上调是通过激活蛋白激酶 RNA 样内质网激酶/真核生物起始因子 2α(eIF2α)分支的内质网应激反应而不是 HIF-1α 信号介导的。转录因子 eIF2α 可以以细胞类型特异性的方式在转录上上调 SLC7A11 的表达或在转录后稳定 SLC7A11 mRNA(62, 82)。此外,p53 可以在未受压力和受压力的条件下(例如,电离辐射和 H2O2)在几种癌细胞中转录上调线粒体谷氨酰胺酶(GLS2)的表达(36)。通过 p53 激活或异位过表达增强 GLS2 的表达促进了谷氨酰胺分解,导致细胞内谷氨酸和 GSH 水平增加(36)。
代谢产物乳酸和富马酸也是 GSH 合成的调节剂。例如,通过抑制单羧酸转运蛋白 1 阻断乳酸出口,增加了人 Raji Burkitt 淋巴瘤细胞中的细胞内乳酸水平,并降低了细胞内 γ-谷氨酰半胱氨酸和 GSH 水平(16)。从机制上讲,乳酸积累抑制了葡萄糖摄取和糖酵解,导致细胞 ATP 水平消耗殆尽。由于 GSH 合成酶 GCL 和 GS 都需要 ATP 才能活动,因此乳酸通过 ATP 消耗抑制了它们的活性,从而抑制了 GSH 的合成(16)。
此外,通过删除延胡索酸水合酶(FH)导致的延胡索酸慢性积累会形成延胡索酰-谷胱甘肽(succinic GSH),这是延胡索酸和GSH的共价加合物,与肾细胞中的持续氧化应激和细胞衰老相关(121)。有趣的是,FH的丢失也通过上调胱氨酸转运蛋白SLC7A11表达以及增加胱氨酸摄取和依赖于谷氨酰胺分解的GSH生物合成,触发了GSH水平的补偿性上升(121)。此外,来自糖尿病大鼠和培养成熟的脂肪细胞的证据表明,作为亲电子体的延胡索酸可以直接与GSH和蛋白质中的半胱氨酸残基通过迈克尔加成反应形成S-(2-琥珀酰)-半胱氨酸(琥珀酰化),支持了延胡索酸可以不可逆地修饰GSH的观点(1, 67)。综合这些证据突出表明,GSH生物合成受到多种相互关联的机制影响,包括转录、转录后和代谢调节。
细胞内GSH/GSSG对的分布
由于与其他氧化还原对相比浓度较高,GSH/GSSG对是细胞的主要氧化还原缓冲剂(88)。细胞内GSH分布在不同的细胞器中(图5)。大多数细胞内GSH(>80%)位于细胞质中,估计浓度为1–11 mM;线粒体GSH占GSH总量的10%-15%,其浓度估计为5–11 mM。细胞内核中的GSH占总量的5%到10%,其浓度与细胞质中的GSH相同或更高;只有少量GSH位于内质网中(55, 88)。细胞内GSH主要以还原形式存在(>98%)。细胞质中的GSH/GSSG比例通常≥30:1–100:1,氧化还原电位为-290 mV;线粒体中的这一比例估计>100:1,氧化还原电位≤ -300 mV。相比之下,内质网中的这一比例似乎为1:1到3:1,氧化还原电位为-175至-185 mV,这解释了内质网相对较为氧化的环境(9, 25, 75, 88)。
值得注意的是,细胞质中的GSH可以与其他细胞内隔室交换。这种隔室交换机制对于线粒体的氧化防御至关重要,因为线粒体无法合成GSH(54)。GSH是一个带负电的分子,不能自由穿越线粒体膜。因此,细胞质中的GSH重定位到线粒体需要特定的载体(图5)。α-酮戊二酸载体(由SLC25A11编码)和二羧酸转运机制(二羧酸载体,由SLC25A10编码)是将细胞质中的GSH导入线粒体的两种潜在候选者,主要在肾脏和肝脏中;三羧酸载体(由SLC25A1编码)是大脑和星形胶质细胞中潜在的GSH转运蛋白(9, 79)。相比之下,某些细胞器,如细胞核,拥有自己独立的GSH池,与细胞质中的GSH无关(88)。这一有趣的现象意味着细胞可能保留核内的GSH以维持还原环境并防止潜在的氧化性DNA损伤。因此,细胞内GSH的分布是器官特异性的,不同池(但不是所有池)之间的交换对于维持GSH稳态至关重要。
哺乳动物细胞可以连续分泌GSH和GSSG到细胞外隔室。在体内,肝脏是循环中GSH的主要来源(55, 88)。健康成年人血浆中的GSH水平估计为2–5 μM,而GSSG浓度则更低(0.14 μM)(40, 66, 101)。有趣的是,老年人和糖尿病患者的血浆GSH水平下降,而GSSG水平相应上升(85),这表明在衰老和糖尿病中向更加氧化的状态转变。如上所述,某些细胞类型表达的GGT酶只能在特定细胞类型的细胞外空间中降解GSH。GGT将GSH水解为谷氨酸和半胱氨酰-甘氨酸,后者进一步被细胞表面的二肽酶降解为半胱氨酸和甘氨酸(2, 102)(图5)。由此产生的氨基酸可以被细胞摄取用于细胞内GSH合成。因此,这种细胞内外通信机制确保在稳态下细胞内的GSH水平得到最佳维持。
四、NAD(P)H和GSH诱导的还原性应激
正如在“细胞氧化还原网络和氧化还原应激”部分讨论的那样,NAD(H)可以调节能量代谢;NAD(P)H和GSH是应对氧化应激不可或缺的还原等效物。矛盾的是,细胞内NAD(P)H和/或GSH的过度积累会导致还原性应激和细胞功能障碍。
NADH和还原性应激
NADH水平的过量会导致还原性应激并最终产生ROS(图6)。线粒体NADH在线粒体呼吸复合物I(NADH脱氢酶)处被氧化为NAD+。越来越多的证据支持,缺氧条件下复合物I处的NADH氧化被抑制,导致NADH积累和随之而来的还原性应激。Berney等人报道,在缺氧条件下,删除两个[NiFe]氢化酶Hyd2和Hyd3使NADH/NAD+比率增加了两倍,导致氧依赖性分枝杆菌中的还原性应激和细胞死亡(6)。同样,缺氧增加了细胞质NADH/NAD+(通过乳酸/丙酮酸的增加表示)并诱导了还原性应激,这与初级大鼠肝细胞中的ATP耗竭和细胞毒性相关(43)。在缺氧条件下,NADH的积累被认为提供了更多的电子用于氧气单电子还原生成O2•−(14)。事实上,在缺氧挑战的牛冠状动脉平滑肌细胞中,细胞质NADH/NAD+和线粒体NAD(P)H水平增加与线粒体ROS产生的增加相耦合(24)。最近,我们报道了缺氧触发了还原性应激并增强了初级人肺成纤维细胞中线粒体ROS的产生(72)。
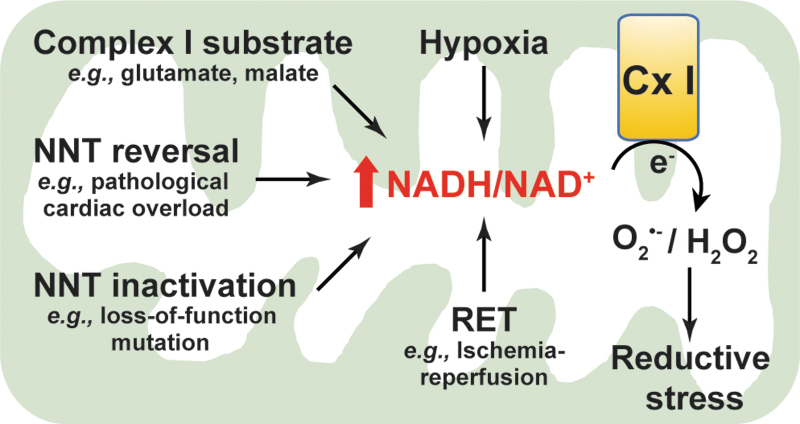
图6.还原性应激由过量的NADH水平引发。 在应激状态下,如外源添加Cx I底物、缺氧、NNT逆转、NNT失活和RET时,线粒体NADH/NAD+比值增加,导致呼吸链复合物I处氧气被单电子还原成O2•−或/和两电子还原成H2O2。大量的还原性ROS水平导致还原性应激,这对细胞功能有害。Cx I, 复合物I;RET, 逆向电子传递。
与这些体外研究一致,在心脏缺血/再灌注(IR)损伤的小鼠模型中,也观察到NADH诱导的还原性应激和线粒体ROS产生增加。在缺血期间,高NADH水平触发了琥珀酸脱氢酶(SDH;复合物II)活性的逆转,导致包括心脏在内的各种组织中琥珀酸积累。再灌注后,积累的琥珀酸被SDH迅速再氧化,通过逆向电子传递模式在复合物I产生ROS,从而引起心脏IR损伤。通过抑制SDH阻断缺血期间琥珀酸积累,减轻了线粒体ROS产生和心脏IR损伤。因此,来自各方面的证据表明,缺氧增加了NADH池,导致ROS产生和还原性应激。
此外,由于高NADH导致的还原性应激也与环境氧张力下的ROS产生增加有关。例如,外源添加复合物I底物(如谷氨酸加苹果酸或α-KG)显著提高了NADH水平和线粒体膜电位,这刺激了分离的大鼠脑线粒体中H2O2的产生约10倍。有趣的是,当通过后续添加ADP或呼吸解偶联剂FCCP增强NADH氧化和线粒体呼吸时,这种增加的H2O2产生减少了50%以上。此外,后续添加复合物I抑制剂鱼藤酮完全阻断了NADH氧化,并在添加FCCP后膜电位崩溃时导致H2O2产生的增强,这表明高NADH水平对于复合物I处的ROS产生至关重要。
同样,在分离的牛心线粒体中,添加NADH(1–3 μM)促进了电子从还原黄素向O2的转移,在几秒钟内(100–200秒)形成复合物I处的O2•−。用抗氧化剂N-乙酰-L-半胱氨酸(NAC;1 mM处理1小时)处理大鼠L6成肌细胞,通过增加NADH/NAD+比率、线粒体H2O2水平和自由基泄漏来诱导还原性应激。此外,胰岛素刺激增加了细胞乳酸产生和乳酸/丙酮酸比例(与NADH/NAD+成正比),这促进了培养的SD大鼠胸主动脉平滑肌细胞中NADH依赖性的NOX酶的细胞O2•−产生。用丙酮酸或草酸预处理显著减轻了这些细胞中的胰岛素诱导的还原性应激。这些无细胞和细胞培养研究表明,高NADH/NAD+促进ROS产生并诱导还原性应激。
线粒体NNT酶催化NADH/NAD+和NADPH/NADP+氧化还原对的可逆转化。越来越多的证据表明,NNT的功能失调导致还原性应激。在C57BL/6J小鼠中,NNT酶因第一外显子的自发突变和多个外显子缺失(外显子7–11)而具有功能丧失的变异。与NNT野生型小鼠(C57BL/6N小鼠)相比,来自C57BL/6J小鼠的分离肝脏线粒体在ADP刺激下具有更高的NADH/总NAD(H)和更低的NADPH/总NADP(H),这与外源性有机过氧化物去除率降低相关。
同样地,在C57BL/6J小鼠的孤立腓肠肌纤维中,加入丙酮酸增加了PDH复合体产生的NADH,这与H2O2水平增加两倍相关(17)。当PDH复合体通量通过丙酮酸和卡尼汀的结合增强时,H2O2的产生进一步增加了五倍(17)。相比之下,当用相同的底物刺激PDH复合体通量时,来自C57BL/6N小鼠(功能性NNT)的孤立肌肉纤维中的H2O2产生并未受到影响(17)。这些结果表明,NNT功能丧失导致还原性应激,通过增加NADH积累和减少NADPH产生,并且NNT可以与PDH复合体整合形成一个调节H2O2产生的氧化还原回路。此外,与对照shRNA转染细胞相比,NNT基因敲低增加了人SK-Hep1细胞在基础和ADP刺激条件下的细胞和线粒体NADH/总NAD(H),而NNT沉默的细胞中细胞和线粒体NADPH/总NADP(H)则明显较低,这与线粒体ROS产生增加相关(33)。这些发现表明,NNT功能丧失阻碍了NADH向NADPH的相互转化,导致NADH积累和还原性应激。
这些观察结果得到了一个体内研究(69)的进一步支持。Nickel等人展示了在由横断主动脉缩窄引起的病理性心脏压力过载下,NNT野生型C57BL/6N小鼠与缺乏功能性NNT活性的C57BL/6J小鼠相比,表现出更严重的心力衰竭和更高的死亡率(69)。野生型小鼠心肌的增强衰竭与NADPH水平的下降以及NADH、H2O2产生和氧化损伤的增加同时发生(69)。这些发现表明,病理性心脏压力过载诱导NNT反向功能模式,以牺牲NADPH为代价产生NADH,导致高NADH/NAD+和还原性应激,进一步促进复合物I处的ROS产生,导致氧化损伤和细胞死亡(图6)。
值得注意的是,NADH/NAD+失衡引起的还原性应激是糖尿病和糖尿病综合症中常见的现象。中心假设是,糖尿病高血糖引起NADH/NAD+比值的伪缺氧增加和还原性应激(57, 103)。具体来说,在慢性高血糖期间,通过糖酵解和多元醇途径增强了NADH的产生,而由于过度激活消耗NAD+的酶(例如,聚ADP核糖聚合酶[PARP]),NAD+水平被耗尽,共同导致NADH/NAD+比值的增加,并通过NADH过载促进线粒体呼吸复合物I的ROS产生,最终导致还原性应激(38, 105, 107)。例如,使用链脲佐菌素诱导的糖尿病大鼠模型,Wu等人发现糖尿病大鼠的肺中多元醇途径的激活和PARP-1的上调与非糖尿病动物相比,这与NADH/NAD+比值的增加、线粒体呼吸复合物I-IV的活性和细胞ROS产生的增加相关(104),支持还原性应激在糖尿病并发症发展中发生的观点。总而言之,这些体外和体内证据强调过量NADH诱导还原性应激并最终促进ROS产生。
NADPH/GSH和还原性应激
NADPH和GSH对于氧化应激防御至关重要;NADPH对于GR回收GSH是必不可少的(参见细胞氧化还原网络和氧化应激部分)。然而,过多的细胞GSH和/或NADPH也会导致还原性应激(图7)。
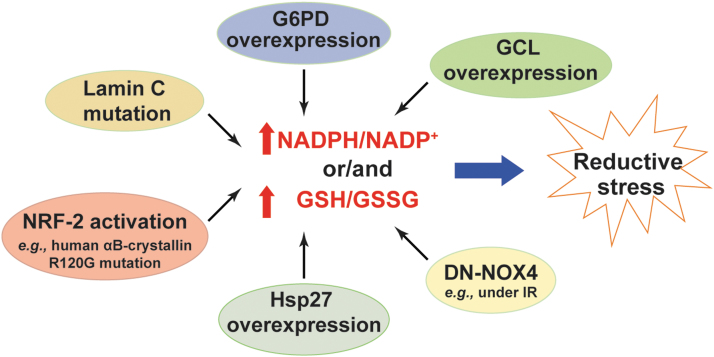
图7还原性应激由NADPH或/和GSH水平过高引发。由于它们的产生增加(例如,G6PD过表达、NRF-2激活、Hsp27过表达、GCL过表达和Lamin C突变)或消耗减少(例如,DN-NOX4过表达)导致NADPH/NADP+或/和GSH/GSSG的增加,从而引起还原性应激。DN-NOX4,显性负性NADPH氧化酶4;Hsp27,热休克蛋白27;IR,缺血/再灌注;NRF-2,核因子(红细胞源性2)样2。
NADPH是NOX酶产生H2O2和O2•−的底物(5)。正如预期的那样,心脏特异性过表达野生型NOX4(WT-NOX4)降低了NADPH/NADP+和GSH/GSSG,这与小鼠在IR挑战下的增强ROS产生和心脏功能障碍相关(116)。相比之下,过表达NOX4的显性负性突变体(DN-NOX4;失去NOX4活性)增加了心肌中这两个氧化还原对的比率。矛盾的是,在IR挑战下的DN-NOX4小鼠也观察到了升高的ROS水平和心脏功能障碍,表明高NADPH和GSH水平也通过尚未确定的机制增强了ROS产生并诱导了还原性应激(116)。
G6PD是细胞质NADPH库的主要来源(参见NAD(H)和NADP(H)对的代谢来源和细胞分布部分)。G6PD过表达增加了细胞内NADPH水平并上调了NOX辅因子的表达,这增强了小鼠胰腺β细胞和胸腺淋巴瘤细胞中的ROS产生和氧化损伤(48, 97)。相比之下,许多疾病模型中发现G6PD活性丧失通过限制高NADPH水平和还原性应激的产生而有益。例如,G6PD缺陷降低了NADPH水平并显著抑制了血管紧张素II(Ang II)诱导的O2•−产生和主动脉中层增厚(64)。在起搏诱导的心力衰竭中观察到了类似的效果,其中抑制G6PD活性消除了衰竭心脏中NADPH水平和ROS产生的升高(28)。
此外,心脏特异性过表达突变的人类αB-晶体蛋白(R120G突变体)的小鼠发展出蛋白质聚集性心肌病,并在心脏中表现出还原性应激,证据是GSH水平、GSH/GSSG以及GR、G6PD、过氧化氢酶和GPx活性的增加(77)。值得注意的是,这些病理改变和还原性应激通过用低表型的G6PD突变体替换R120G小鼠中的野生型G6PD得到了显著正常化(77),这表明G6PD介导的还原性应激有助于这种疾病的发展。机制上,R120G突变体小鼠中观察到的还原性应激归因于核因子(红细胞源性2)样2(NRF-2)信号通路的激活,该通路转录上调了关键抗氧化酶(例如,GR、G6PD、GPx1和过氧化氢酶)和GSH合成酶(例如,GCLC和GCLM)的mRNA或/和蛋白质表达,导致心脏组织中GSH含量和GSH/GSSG比值的增加,以及ROS水平的降低(77, 91)。重要的是,敲除NRF2显著中止了基因表达的上调,正常化了还原性应激,并减少了R120G突变体小鼠的心脏肥大(41, 78)。
同样地,果蝇肌肉组织中的Lamin C突变或人类肌营养不良症患者导致了还原性应激,表现为GSH和NADPH水平的增加以及NRF-2途径的激活(15)。在这个模型中,NADPH水平的增加是由IDH活性升高贡献的,但与G6PD和6PGD活性无关(15)。与氧化性应激下NRF-2激活是通过其隔离蛋白Keap-1的氧化失活介导的不同,还原性应激下的激活是通过上调自噬适配器p62/SQSTM1介导的,它也能结合Keap-1,从而使得NRF-2能够核转运并激活其靶基因(15)。
此外,心脏特异性热休克蛋白27(Hsp27)过表达的转基因小鼠发展出心脏肥大和功能障碍,这与心脏中GSH水平、GSH/GSSG比值和GPx1活性的增加以及总ROS水平的降低相关(119)。抑制GPx1活性部分纠正了Hsp27转基因小鼠的心肌病,这表明由过量GSH水平和GPx1活性引起的还原性应激有助于该模型中的心脏功能障碍(119)。此外,通过补充巯基供体NAC或过表达GSH合成酶GCLC或/和GCLM亚基来增加细胞内GSH水平,降低了细胞内ROS水平,并通过7-12mV降低了细胞2GSH/GSSG还原电势(117)。矛盾的是,这种更还原的氧化还原环境与线粒体氧化和细胞死亡的增加相关,这表明升高的GSH水平导致还原性应激和细胞毒性(117)。综合这些证据清楚地支持过量NADPH或/和GSH水平诱导还原性应激。
五、还原性应激的代谢反应
NAD(H)、NADP(H)和GSH/GSSG氧化还原对是细胞代谢的关键调节器。具体来说,糖酵解和TCA循环产生NADH,为线粒体OXPHOS和ATP产生提供电子(56)。NADP+支持PPP产生对核苷酸、氨基酸和脂质的还原性生物合成不可或缺的NADPH(56)。GSH将氨基酸代谢与细胞氧化还原状态联系起来。因此,这三个氧化还原对比率改变引起的还原性应激影响细胞代谢,反之亦然。
NAD(P)H诱导的还原性应激下的代谢协调
在缺氧条件下,细胞代谢从线粒体OXPHOS转向糖酵解以产生能量,这主要是通过HIF-1α信号激活来介导的。正如在NAD(P)H和GSH引起的还原性应激部分提到的,缺氧也通过NADH积累诱导还原性应激。直到最近,还原性应激与代谢重编程之间是否存在联系尚未被很好理解。
我们和其他研究者展示了,在各种原发性和癌症细胞中,作为对缺氧的基本反应,L-2-羟基戊二酸(L2HG)(α-KG通过MDH1/2或LDHA还原的代谢产物)选择性地积累(39, 72)。L2HG水平的积累与细胞NADH/NAD+的增加和线粒体ROS产生的增加相关,这表明在缺氧条件下存在还原性应激(72)。引人注目的是,通过敲低L2HG脱氢酶(唯一已知将L2HG氧化回α-KG的酶)来增加细胞L2HG水平,降低了线粒体氧消耗和乳酸产生,这表明L2HG可以抑制线粒体呼吸和糖酵解,以减少它们的NADH产生并减轻相关的还原性应激(72)。
线粒体NNT酶是线粒体NADPH水平的重要来源(见NAD(H)和NADP(H)对的代谢来源和细胞分布部分)。NNT过表达增加了NADPH/NADP+,导致黑色素瘤细胞中的还原性应激(23)。有趣的是,依赖NADPH的还原性应激刺激了谷氨酰胺分解产生α-KG,然后通过TCA循环中的氧化脱羧或通过IDH2介导的还原羧化转化为琥珀酰辅酶A或柠檬酸(23)。相比之下,NNT沉默降低了NADH/NAD+和NADPH/NADP+,这与谷氨酰胺分解的下降和葡萄糖氧化流入TCA循环及由此产生的能量的增加相关(23)。这些结果表明,在还原性应激条件下,黑色素瘤细胞将其能源来源从葡萄糖切换到谷氨酰胺。
最近的一项研究还报告了在NNT基因敲低的人SK-Hep1细胞中出现还原性应激和代谢重编程(33)。具体来说,NNT的敲低导致细胞和线粒体NADH/总NAD(H)升高,细胞和线粒体NADPH/总NADP(H)降低,同时伴随着线粒体O2•−产生的增加,表明NNT沉默在这些细胞中诱导了还原性应激(33)。有趣的是,NNT沉默还促进了线粒体膜电位超极化,增加了氧气消耗和ATP产生,并减少了乳酸产生,这意味着为了纠正这些细胞中NADH的积累,代谢从糖酵解转向了线粒体OXPHOS。这种代谢转变使这些细胞对线粒体OXPHOS抑制剂鱼藤酮的细胞毒性效应更加敏感,但对糖酵解抑制剂3-溴丙酮酸更加抵抗(33)。此外,在NNT沉默的细胞中也观察到增强的谷氨酰胺分解和还原性羧化作用,导致α-KG及其衍生代谢物(如柠檬酸、富马酸和苹果酸)的积累。从机制上讲,这种对糖酵解活性的抑制是由于HIF-1α蛋白的不稳定,可能是通过α-KG依赖的PHD酶介导的羟化作用和HIF-1α蛋白的增强降解(33)。
此外,从C57BL/6J小鼠分离的原代小鼠主动脉内皮细胞与来自C57BL/6N小鼠的细胞相比,当受到Ang II刺激时,消耗的氧气更少,GPx活性更低,产生的O2•−更多,这表明NNT功能丧失损害了线粒体功能(49)。此外,越来越多的证据强烈支持在缺乏功能性NNT的C57BL/6J小鼠中观察到的还原性应激与系统代谢改变相关,表现为葡萄糖耐量不良、葡萄糖诱导的胰岛素分泌和能量消耗减少,以及对高脂饮食诱导的肥胖的易感性增加(17, 20, 21, 33, 68, 80)。
值得注意的是,NNT也可以以反向模式运作,从NADPH和NAD+生成NADH,导致NADH/NAD+升高、还原性应激和代谢适应。例如,在用横断面主动脉缩窄术挑战的C57BL/6N小鼠中,NNT通过其反向酶促模式运作,这促进了NADH的产生,以牺牲NADPH和其抗氧化能力为代价(69)。增加的NADH产生通过加速线粒体OXPHOS来促进线粒体呼吸,这与NADPH的抗氧化能力受损相结合,导致了线粒体ROS产生和氧化损伤的增加,最终导致心脏功能障碍(69)。这些发现表明,心肌细胞通过刺激线粒体呼吸的氧化来纠正NADH诱导的还原性应激。
在C57BL/6N小鼠的分离胰岛中,葡萄糖刺激增加了NADPH/总NADP(H),这与线粒体GSH氧化的减少相关(86)。有趣的是,增加的比例是由NNT酶的反向操作的抑制而不是其正向操作的刺激引起的,因为相同的挑战也提高了NADH/总NAD(H)。NAD(P)H水平的上升与葡萄糖刺激的胰岛素分泌、糖酵解(如通过提高的G-6-P和F-6-P水平所示)和线粒体呼吸(如通过增加的葡萄糖氧化、氧气消耗、ATP产生以及TCA循环中间代谢物柠檬酸、苹果酸和富马酸的水平所示)的增强相关(86)。总的来说,NNT因此是连接细胞代谢和还原性应激的关键酶。
正如在NAD(H)和NADP(H)对的代谢来源和细胞分布部分所讨论的,苹果酸/天冬氨酸穿梭是一个关键机制,用于维持细胞质中高NAD+水平和线粒体中高NADH水平的隔室交换。这种交换机制的破坏可能会扰乱两个隔室中的NADH/NAD+,从而可能影响细胞氧化还原平衡和能量代谢(图8)。例如,敲低线粒体谷氨酸-草酰乙酸转氨酶(GOT2),苹果酸/天冬氨酸穿梭中的一个关键酶,增加了细胞质中的NADH水平,并同时降低了线粒体中的NADH水平,这与NADPH/NADP+的降低和PANC-1细胞中细胞ROS产生的增加相伴(109)。GOT2的沉默还显著抑制了细胞ATP产生和细胞增殖,表明抑制了线粒体呼吸(109)。有趣的是,通过重新表达一个乙酰化模拟的GOT2恢复GOT2活性,在这些细胞中抑制了细胞ROS的产生,并恢复了线粒体NADH水平、细胞ATP产生和细胞增殖(109)。
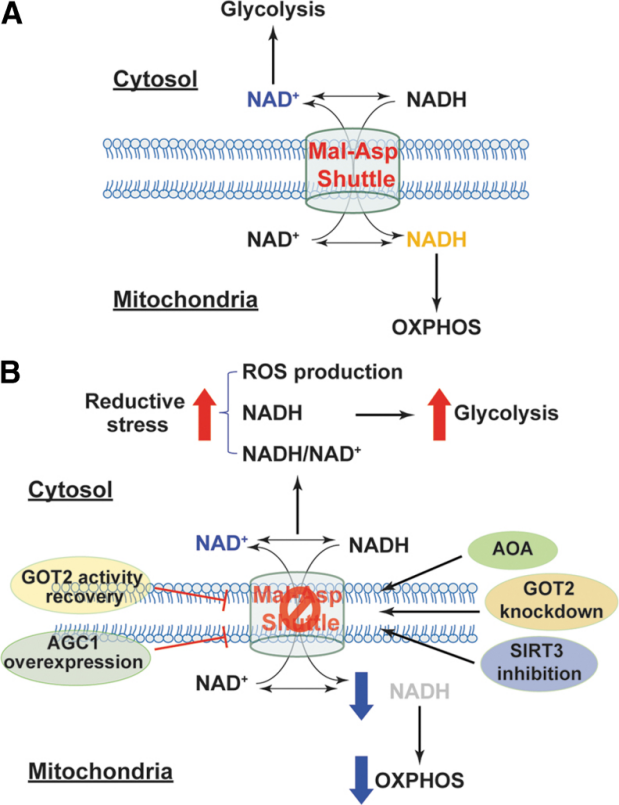
图8. 苹果酸/天冬氨酸穿梭的抑制诱导还原性应激和代谢重编程。(A)苹果酸/天冬氨酸穿梭交换细胞质中的NAD(H)与线粒体中的NAD(H),以维持高细胞质NAD+水平(糖酵解所需)和高线粒体NADH水平(为线粒体氧化磷酸化提供电子)。(B)通过沉默GOT2、抑制SIRT3活性或使用化学抑制剂AOA来抑制苹果酸/天冬氨酸穿梭,导致细胞质NADH的积累以及细胞质NADH/NAD+的增加和还原性ROS的产生,表明出现了还原性应激。NADH诱导的还原性应激将细胞代谢从线粒体呼吸转移到糖酵解。通过AGC1过表达或恢复GOT2活性增强该穿梭活性,显著增加线粒体NADH水平和增强线粒体呼吸。AGC1,天冬氨酸-谷氨酸载体1;AOA,氨基氧乙酸;GOT2,谷氨酸-草酰乙酸转氨酶;Mal-Asp shuttle,苹果酸/天冬氨酸穿梭;OXPHOS,氧化磷酸化;SIRT3,去乙酰化酶家族成员3。
此外,用氨基氧乙酸(AOA,一种强力的天冬氨酸转氨酶抑制剂)抑制苹果酸/天冬氨酸穿梭,增加了细胞质NADH/NAD+,反映在初级猪主动脉平滑肌细胞的细胞质部分中乳酸/丙酮酸和甘油-3-磷酸/DHAP的升高,导致细胞质室的还原环境更加强烈(3, 4)。值得注意的是,AOA处理还减弱了葡萄糖氧化和氧气消耗,但增加了糖酵解活性和乳酸产生,表明代谢向糖酵解转移(3, 4)。这些结果表明,苹果酸/天冬氨酸穿梭的干扰导致细胞质中NADH的保留和还原性应激,减少线粒体中NADH的可用性,并可能增加细胞质中NAD+水平,从而导致NADH依赖性线粒体呼吸的抑制和NAD+依赖性糖酵解的增强。
这种穿梭的重要角色也在体内得到了证明。心脏特异性删除线粒体复合物I亚基NDUFS4蛋白导致小鼠心脏线粒体中NADH的积累和NADH/NAD+的升高,这与病理性压力超负荷或异丙肾上腺素诱导的心脏衰竭加速发展相关(42, 47)。有趣的是,来自这些小鼠的分离心肌细胞比野生型小鼠的细胞产生的H2O2和O2•−更少(42, 47),表明复合物I功能障碍导致衰竭心脏中的还原性应激。由于超生理水平的NADH被报道抑制NAD+依赖性的去乙酰化酶(SIRT1–7)(50),线粒体中积累的NADH抑制了线粒体SIRT3活性,导致线粒体蛋白的超乙酰化(42, 47)(图8)。具体来说,苹果酸/天冬氨酸穿梭蛋白MDH2、α-KG/苹果酸载体和GOT2被发现超乙酰化,导致细胞质NADH在线粒体中的转运和氧化受到抑制,这与氧气消耗和心脏能量学(通过磷酸肌酸/ATP的减少表现出来)的减少相关(42, 47)。所有这些病理和生化变化都通过给小鼠喂食NAD+前体NAM单核苷酸或在心脏组织中超表达NAM磷酸核糖基转移酶(NAD+生物合成的限速酶)而被显著改善(42, 47)。因此,这些结果支持高NADH/NAD+诱导的还原性应激损害心脏的线粒体功能和能量代谢。
相比之下,过表达人天冬氨酸-谷氨酸载体1(AGC1,由SLC25A12基因编码)在大鼠INS-1E β细胞中增强了葡萄糖刺激下的苹果酸/天冬氨酸穿梭活性,如线粒体NAD(P)H水平和细胞谷氨酸水平的增加所示(83)(图8)。此外,与空腺病毒载体转染的细胞相比,AGC1过表达还增强了葡萄糖诱导的葡萄糖氧化、线粒体膜电位超极化、ATP产生和胰岛素分泌的增加,但抑制了乳酸分泌(83),表明线粒体中提高的NAD(P)H水平促进了线粒体呼吸和能量产生。这些证据一致支持苹果酸/天冬氨酸穿梭对于将细胞质NADH的电子传递到线粒体进行氧化磷酸化至关重要,而这种穿梭的中断导致还原性应激和代谢功能障碍。
在GSH诱导的还原性应激下的代谢协调
正如在GSH生物合成和细胞分布部分讨论的,谷氨酸是GSH的前体氨基酸。谷氨酸也是α-KG的来源,通过谷氨酰胺分解,其中谷氨酰胺被GLSs(细胞质GLS1和线粒体GLS2)水解成谷氨酸,然后谷氨酸被GLUDs转化为α-KG。因此,增强谷氨酰胺分解可以增加细胞GSH水平并改变细胞氧化还原状态和能量代谢。例如,三种人类癌细胞系中过表达GLS2显著提高了细胞谷氨酸、GSH和NADH水平以及GSH/GSSG比例,这与细胞基础ROS水平的降低相关,意味着这些细胞发生了还原性应激。有趣的是,过表达GLS2的细胞显示出细胞α-KG水平、线粒体氧气消耗和ATP产生的增加,表明在还原性应激下线粒体氧化磷酸化和TCA循环得到增强。重要的是,通过沉默GLS2显著正常化了还原性应激和代谢刺激(36)。
相比之下,GLS1而不是GLS2被发现介导了在小鼠骨骼肌细胞中HIF-1α对谷氨酰胺分解的增强作用。在常氧条件下,通过PHD2缺失激活HIF-1α信号增强了谷氨酰胺的摄取并上调了GLS1表达,导致谷氨酰胺分解增强和细胞GSH水平以及GSH/GSSG比例的增加,这与抗氧化基因(SOD1、SOD2、过氧化氢酶、GPx1和GR)的上调以及细胞和线粒体ROS产生的减少相关,这是还原性应激的指标(96)。有趣的是,这种还原性应激伴随着氧气消耗和棕榈酸β-氧化的减少以及糖酵解活性和糖原储存的增加(96),表明线粒体功能的抑制和糖酵解及糖原生成的刺激。
此外,最近的一项研究揭示了在炎症期间GSH对小鼠T细胞中的代谢整合和重编程的必要性(61)。在激活时,小鼠T细胞表达的GCLC mRNA比静息T细胞高出6-10倍,这伴随着细胞GSH水平的升高。激活的T细胞通过增强谷氨酰胺进入TCA循环和葡萄糖进入糖酵解的通量,以c-MYC依赖的方式重编程它们的代谢(61)。这些代谢适应性反应在GCLC缺失的T细胞中被有效消除,并通过外源性添加GSH或其前体NAC重新复制(61)。这些发现总体上强调了GSH相关的还原性应激与能量代谢之间的密切联系。
六、结论
作为结束语,NAD(H)、NADP(H)和GSH/GSSG氧化还原对是许多氧化还原和代谢酶的辅因子或/和底物。在每个氧化还原对内,还原形式和氧化形式之间的微妙平衡是支持细胞氧化还原平衡和能量代谢的先决条件。当这种平衡丧失时,细胞内NAD(P)H和GSH水平的过量会导致还原性应激、代谢应激和细胞功能障碍。
作为一个新兴概念,还原性应激复杂化了我们对细胞氧化还原环境的理解,但扩展了我们的认识。传统上,还原环境被认为对细胞功能和生物过程有益,因为氧化环境可能导致蛋白质、脂质和核酸的氧化损伤。还原性应激强调了过量的还原等效物(NAD(P)H和GSH)也对细胞有害的事实,包括通过许多机制,包括破坏ROS的信号功能、诱导ROS的产生、扰乱细胞代谢和抑制蛋白质二硫键的形成。因此,哺乳动物细胞必须维持一个平衡的氧化还原环境,以避免氧化和还原损伤。
关于还原性应激的潜在机制及其生物学后果以及细胞如何响应还原性应激的知识仍然有限。例如,尽管很明显缺氧下NADH的积累会诱导还原性应激,但尚不清楚(i)在缺氧下NADH是如何积累的,除了通过线粒体呼吸的氧化抑制;(ii)NADH的积累是否是 compartment-specific;(iii)两种NADH穿梭是否参与重新分配;(iv)NNT酶是否参与重新分配;(v)NADH的积累如何影响葡萄糖、脂质和谷氨酰胺的代谢命运;以及(vi)细胞如何响应以减轻这种伤害。这些问题的答案在很大程度上仍然是开放的,需要未来的实验努力。
此外,GSH诱导的还原性应激对细胞代谢的影响以及反之亦然也是未解决的问题。这项工作总结了文献中可用的有限信息,关于调节GSH生物合成如何影响细胞代谢。值得注意的是,GSH调节细胞代谢的另一个重要机制是通过蛋白质S-谷胱甘肽化。像磷酸化一样,S-谷胱甘肽化是一种重要的翻译后修饰机制,调节目标蛋白的生物学功能、结构蛋白折叠和亚细胞定位。蛋白质可以通过非酶促和酶促反应进行谷胱甘肽化。
前者类型的S-谷胱甘肽化主要在氧化应激期间进行,通常是非特异性和不可逆的(58, 79)。ROS将GSH氧化成GSSG,后者与蛋白质上的半胱氨酸残基(-SH)反应形成PSSG。充分的证据表明,在氧化应激下,代谢酶容易发生谷胱甘肽化。例如,糖酵解酶(如GAPDH、丙酮酸激酶、醛缩酶、磷酸甘油酸激酶、三磷酸异构酶)、TCA循环酶(即乌头酸酶、KGDH、IDH3、琥珀酰辅酶A转移酶)和线粒体氧化磷酸化蛋白复合物(复合物I-V)可以被谷胱甘肽化,这会导致它们的酶活性的激活或失活,从而调节细胞能量学(12, 18, 25, 29, 44, 59, 70)。
相比之下,酶驱动的S-谷胱甘肽化通常是特异性和可逆的,并且受到局部GSH/GSSG池波动的高度控制(58, 79)。谷胱甘肽-S-转移酶是主要的介导S-谷胱甘肽化的酶,通过将GSH添加到蛋白质的半胱氨酸残基产生PSSG(98);而Grx1和Grx2是催化可逆反应的主要酶(58)。到目前为止,关于在还原性应激下(高GSH/GSSG)蛋白质特别是代谢酶是否可以发生谷胱甘肽化,以及这种修饰如何损害细胞代谢活动,知之甚少。这些研究主题值得未来的研究。
https://blog.sciencenet.cn/blog-41174-1433721.html
上一篇:衰老的时钟是如何运转
下一篇:氢气防治肾脏疾病研究进展【综述】